

Pre-clinical data supporting immunotherapy for HIV using CMV-HIV-specific CAR T cells with CMV vaccine
- PMID: 35573050
- PMCID: PMC9062763
- DOI: 10.1016/j.omtm.2022.04.007
Pre-clinical data supporting immunotherapy for HIV using CMV-HIV-specific CAR T cells with CMV vaccine
Abstract
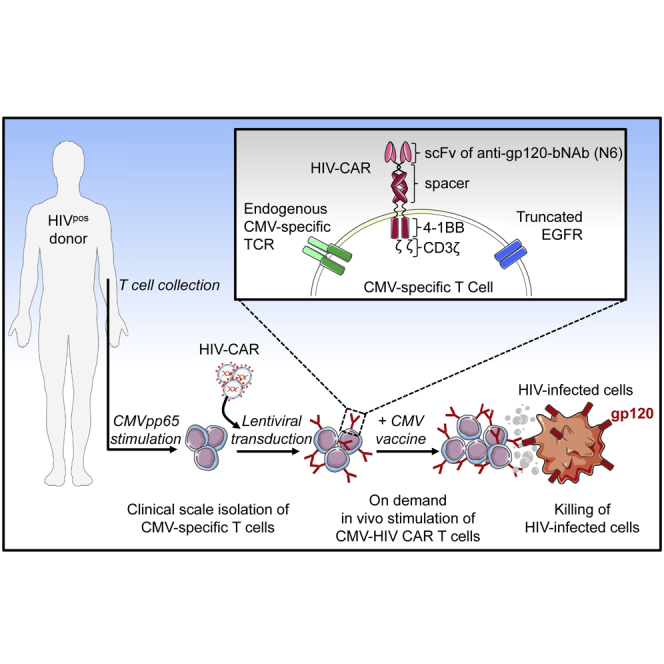
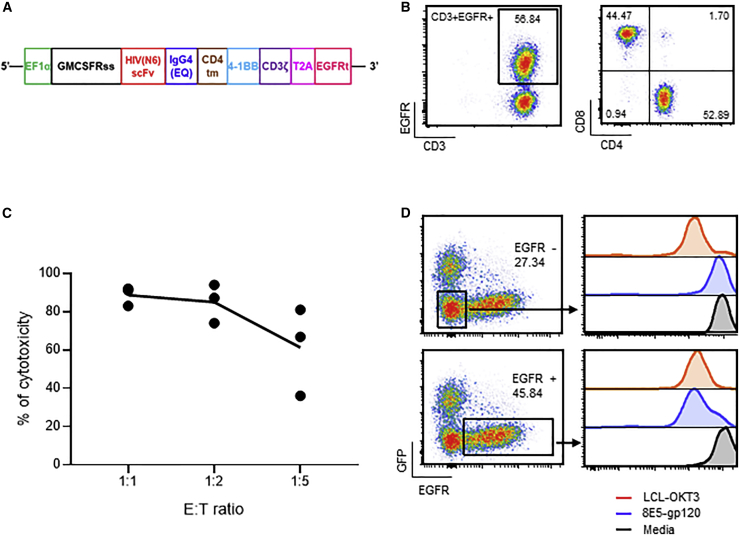
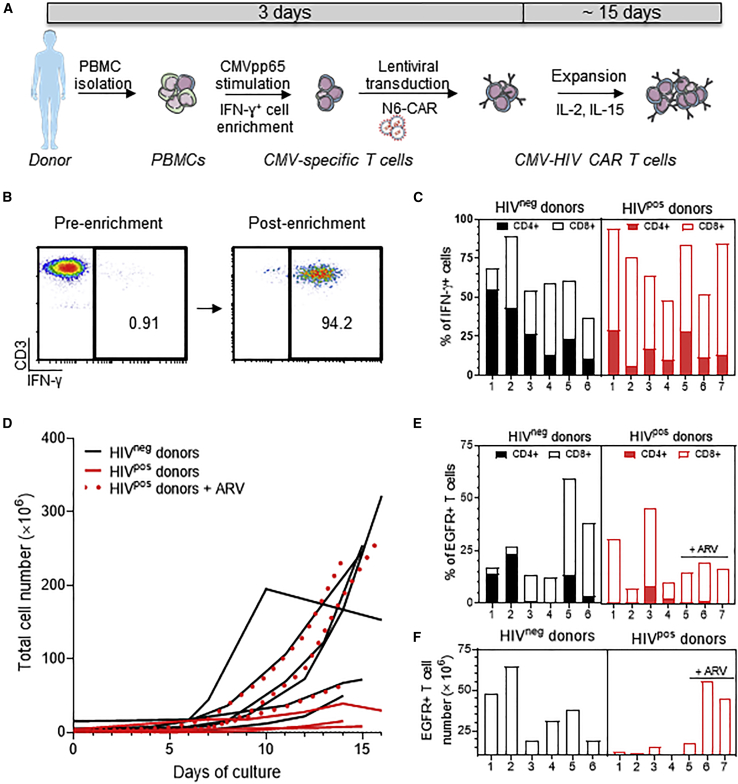
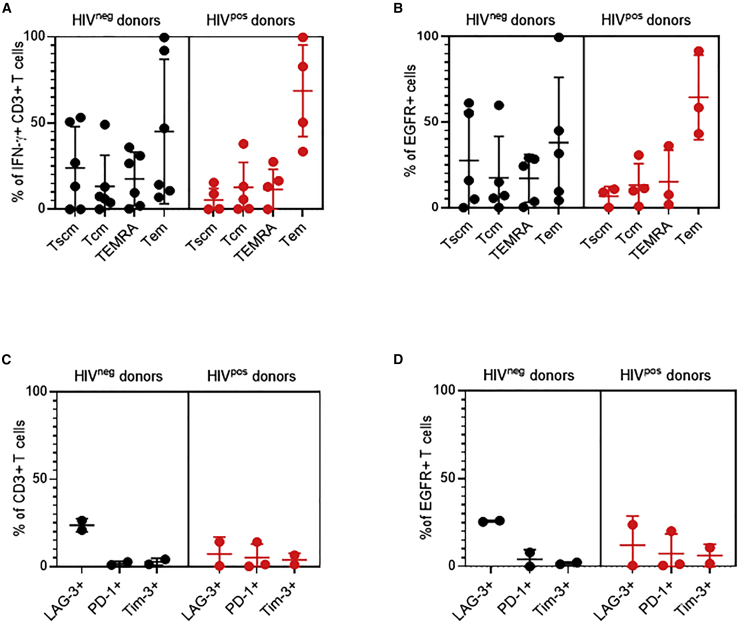
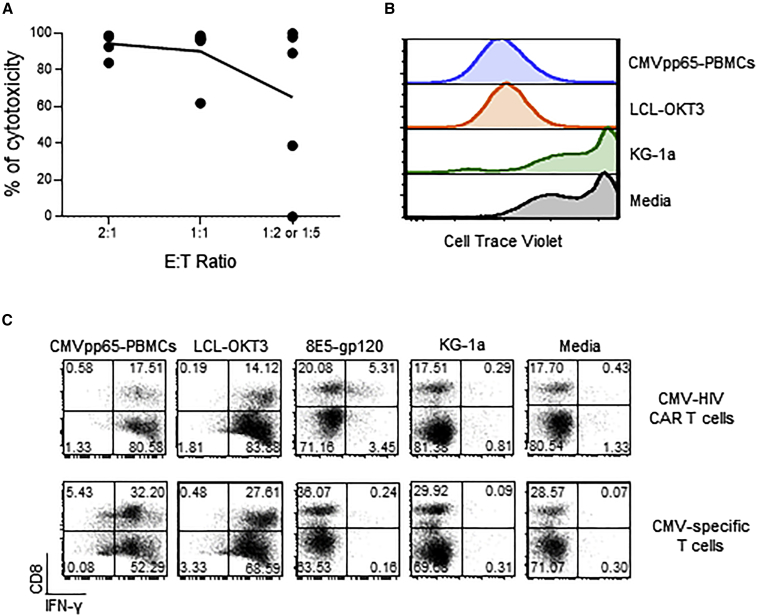
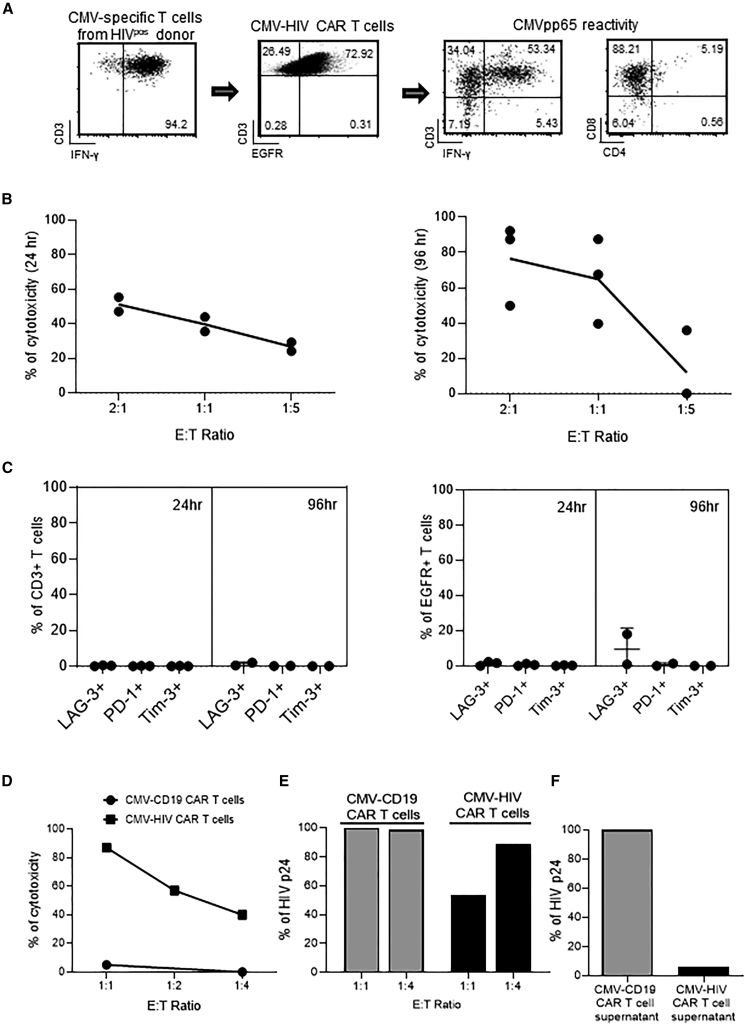
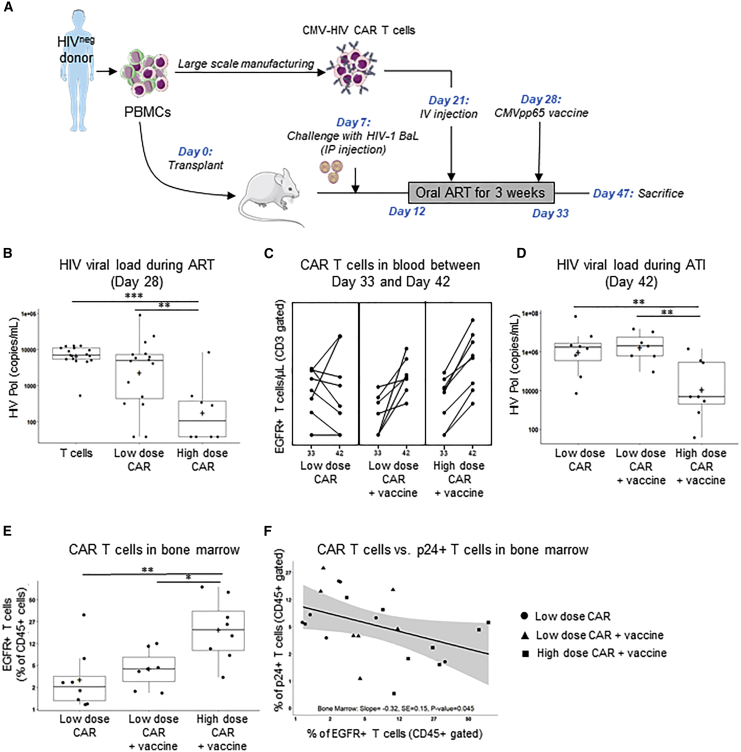
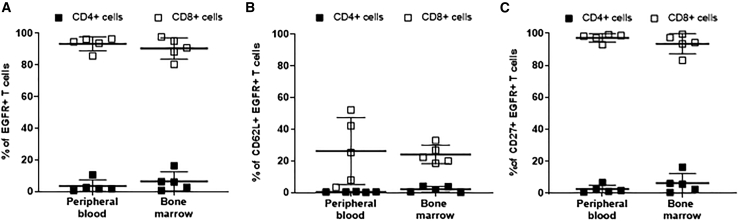
T cells engineered to express HIV-specific chimeric antigen receptors (CARs) represent a promising strategy to clear HIV-infected cells, but to date have not achieved clinical benefits. A likely hurdle is the limited T cell activation and persistence when HIV antigenemia is low, particularly during antiretroviral therapy (ART). To overcome this issue, we propose to use a cytomegalovirus (CMV) vaccine to stimulate CMV-specific T cells that express CARs directed against the HIV-1 envelope protein gp120. In this study, we use a GMP-compliant platform to engineer CMV-specific T cells to express a second-generation CAR derived from the N6 broadly neutralizing antibody, one of the broadest anti-gp120 neutralizing antibodies. These CMV-HIV CAR T cells exhibit dual effector functions upon in vitro stimulation through their endogenous CMV-specific T cell receptors or the introduced CARs. Using a humanized HIV mouse model, we show that CMV vaccination during ART accelerates CMV-HIV CAR T cell expansion in the peripheral blood and that higher numbers of CMV-HIV CAR T cells were associated with a better control of HIV viral load and fewer HIV antigen p24+ cells in the bone marrow upon ART interruption. Collectively, these data support the clinical development of CMV-HIV CAR T cells in combination with a CMV vaccine in HIV-infected individuals.
Keywords: HIV/AIDS; N6; broadly neutralizing antibody (bNAb); chimeric antigen receptor T cell (CAR T); cytomegalovirus (CMV) vaccine; immunotherapy.
Conflict of interest statement
Competing interests: A patent associated with this study covering the work has been held and submitted by City of Hope (US2016/024560) with X.W. and S.J.F. as inventors who could potentially receive licensing royalties in the future. The remaining authors declare no competing interests.
Figures
References
-
- Günthard H.F., Saag M.S., Benson C.A., del Rio C., Eron J.J., Gallant J.E., Hoy J.F., Mugavero M.J., Sax P.E., Thompson M.A., et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2016 recommendations of the international antiviral society-USA panel. JAMA. 2016;316:191–210. doi: 10.1001/jama.2016.8900. - DOI - PMC - PubMed
-
- Patel S., Hanajiri R., Grant M., Saunders D., Van Pelt S., Keller M., Hanley P.J., Simon G., Nixon D.F., Hardy D., et al. HIV-specific T cells can Be generated against non-escaped T cell epitopes with a GMP-compliant manufacturing platform. Molecular therapy. Methods Clin. Dev. 2020;16:11–20. doi: 10.1016/j.omtm.2019.10.001. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
