

HDAC6 modulates myofibril stiffness and diastolic function of the heart
- PMID: 35575093
- PMCID: PMC9106344
- DOI: 10.1172/JCI148333
HDAC6 modulates myofibril stiffness and diastolic function of the heart
Abstract
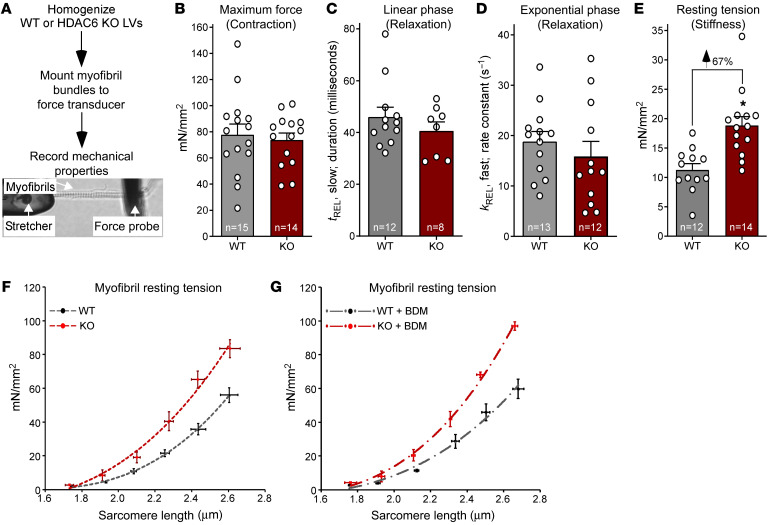
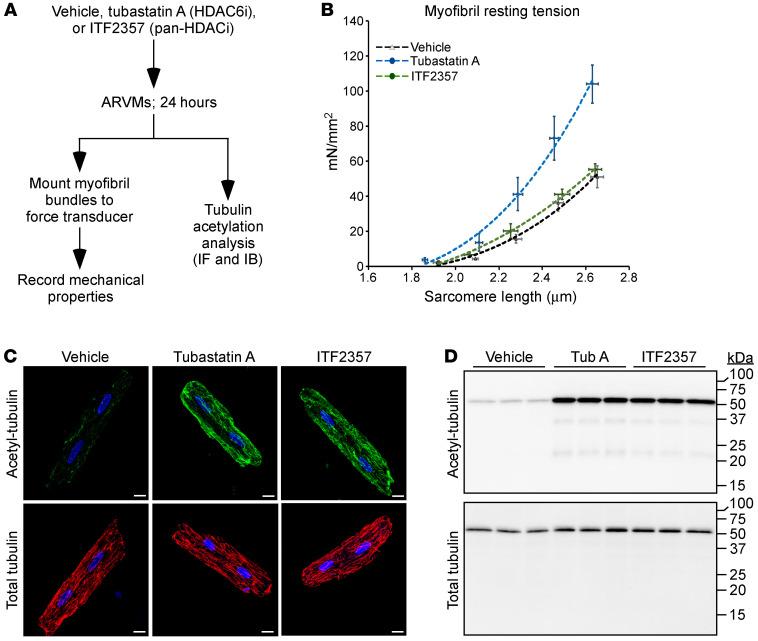
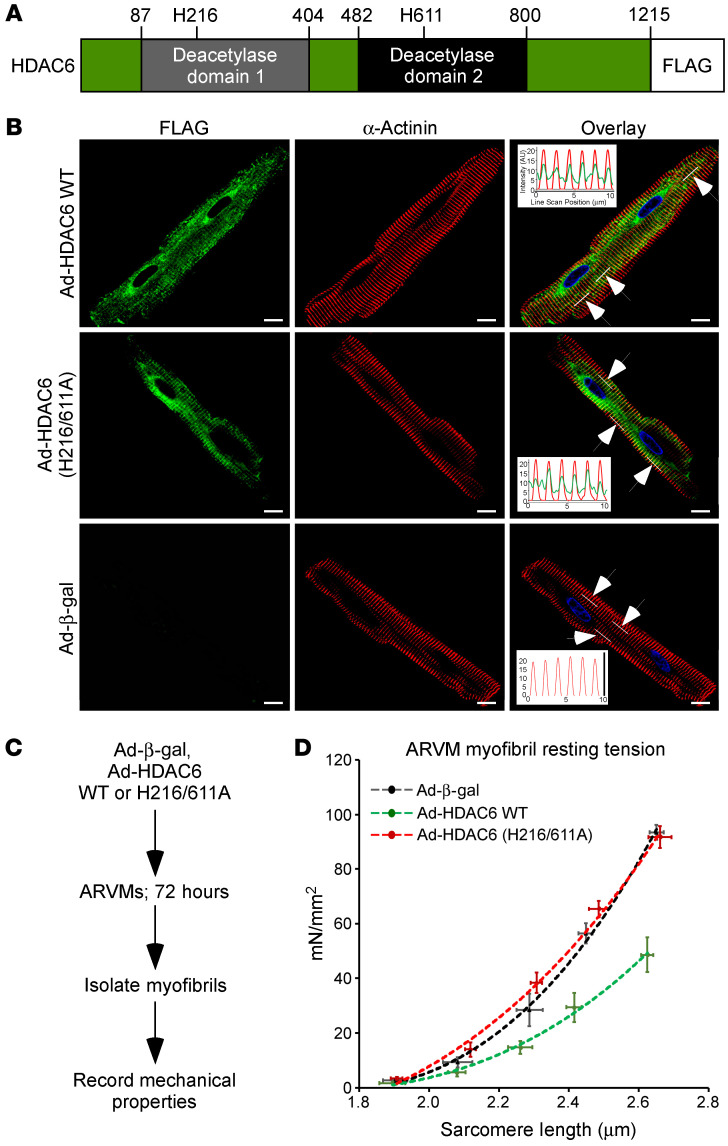
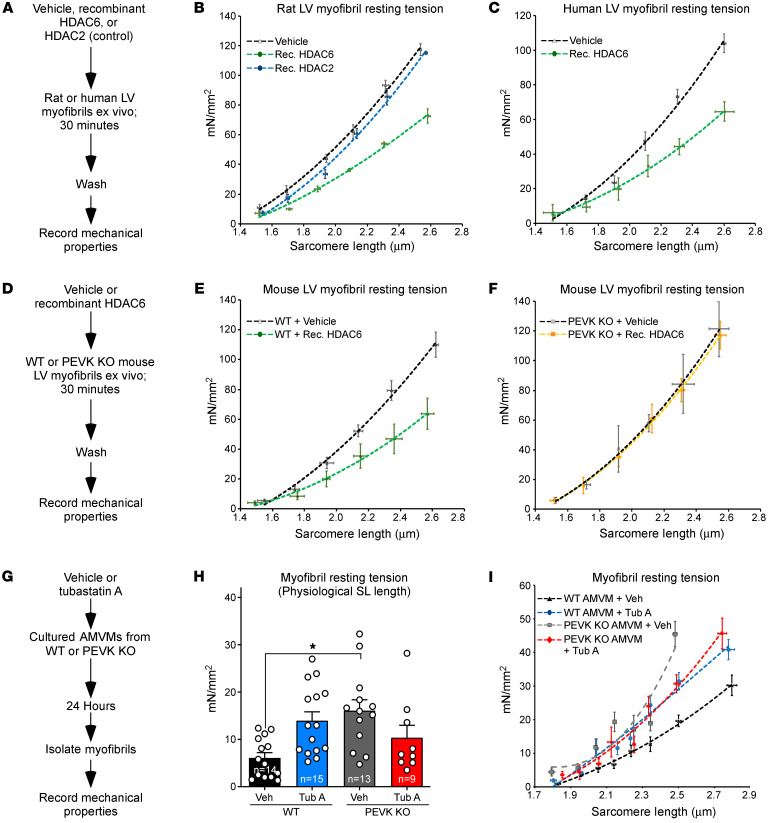
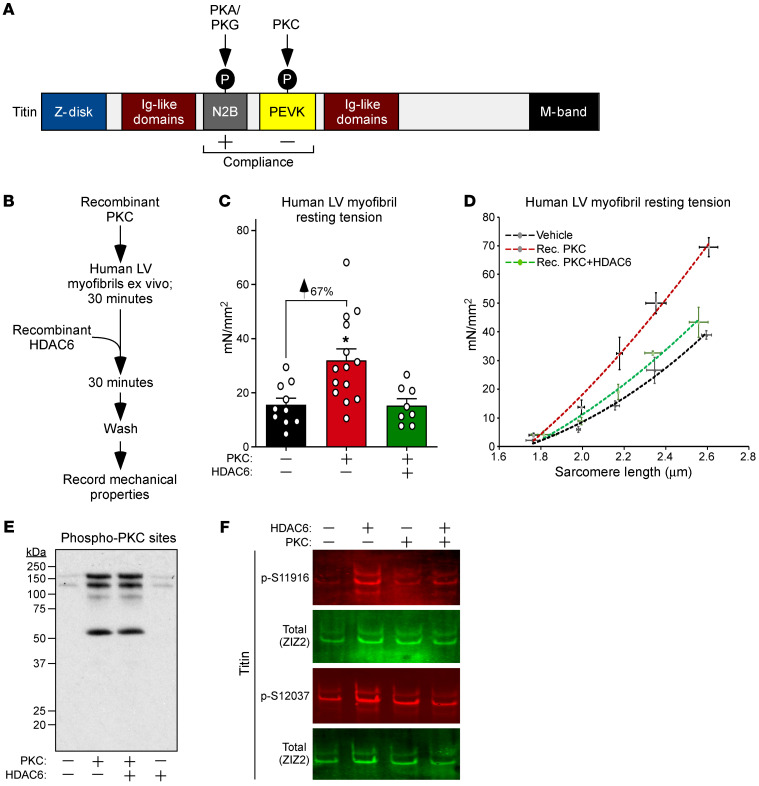
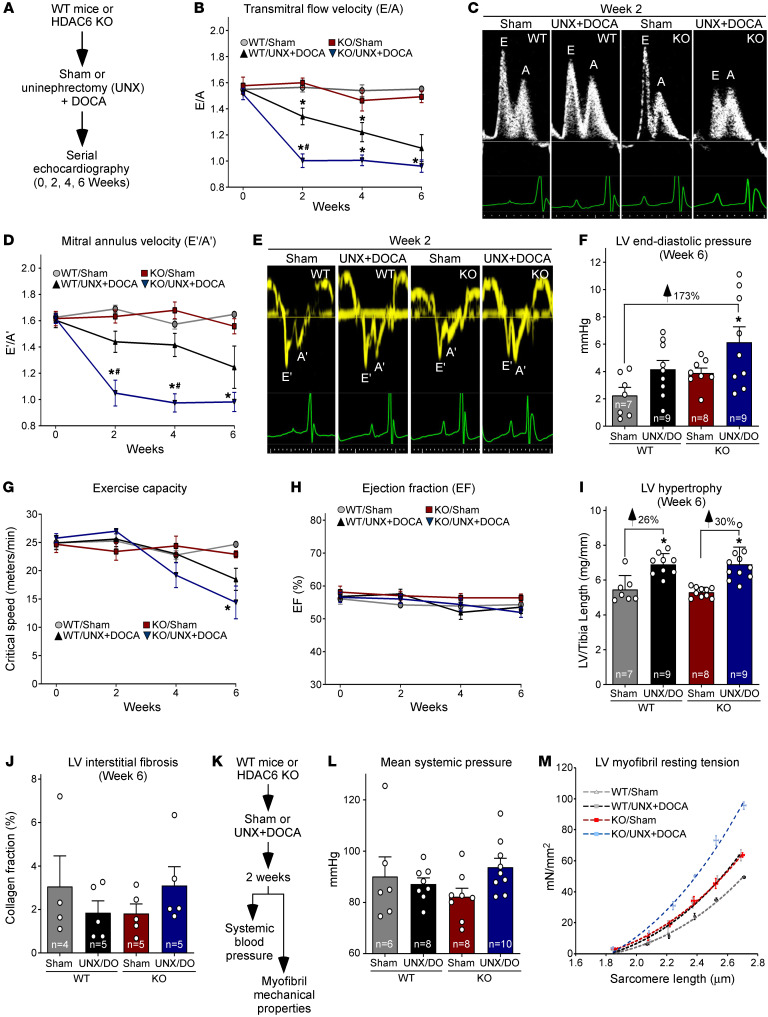
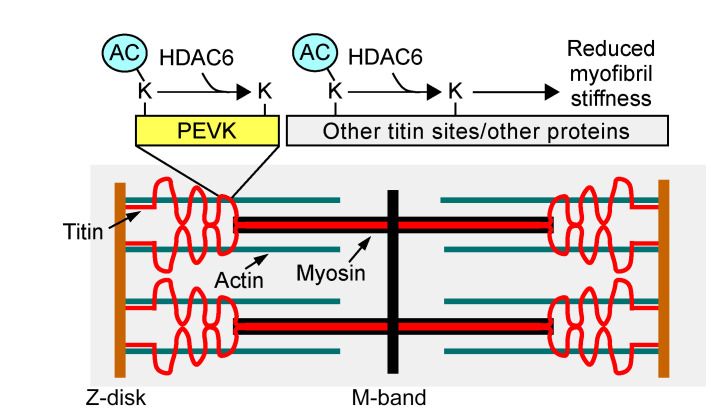
Passive stiffness of the heart is determined largely by extracellular matrix and titin, which functions as a molecular spring within sarcomeres. Titin stiffening is associated with the development of diastolic dysfunction (DD), while augmented titin compliance appears to impair systolic performance in dilated cardiomyopathy. We found that myofibril stiffness was elevated in mice lacking histone deacetylase 6 (HDAC6). Cultured adult murine ventricular myocytes treated with a selective HDAC6 inhibitor also exhibited increased myofibril stiffness. Conversely, HDAC6 overexpression in cardiomyocytes led to decreased myofibril stiffness, as did ex vivo treatment of mouse, rat, and human myofibrils with recombinant HDAC6. Modulation of myofibril stiffness by HDAC6 was dependent on 282 amino acids encompassing a portion of the PEVK element of titin. HDAC6 colocalized with Z-disks, and proteomics analysis suggested that HDAC6 functions as a sarcomeric protein deacetylase. Finally, increased myofibril stiffness in HDAC6-deficient mice was associated with exacerbated DD in response to hypertension or aging. These findings define a role for a deacetylase in the control of myofibril function and myocardial passive stiffness, suggest that reversible acetylation alters titin compliance, and reveal the potential of targeting HDAC6 to manipulate the elastic properties of the heart to treat cardiac diseases.
Keywords: Cardiology; Heart failure.
Conflict of interest statement
Figures
References
-
- Nagueh SF, et al. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110(2):155–162. doi: 10.1161/01.CIR.0000135591.37759.AF. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
