

Cryo-electron Microscopy of Adeno-associated Virus
- PMID: 35575684
- PMCID: PMC9479082
- DOI: 10.1021/acs.chemrev.1c00936
Cryo-electron Microscopy of Adeno-associated Virus
Abstract
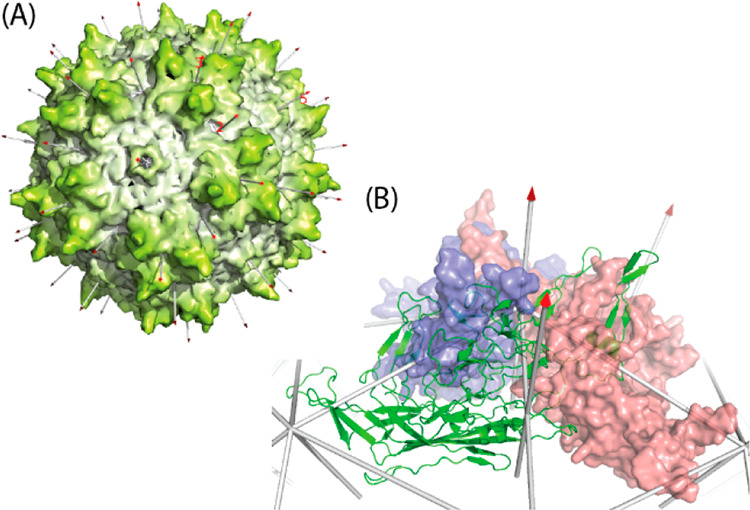
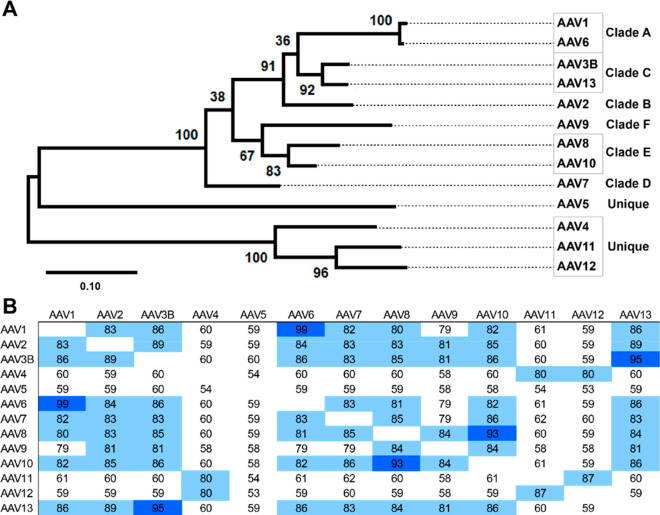
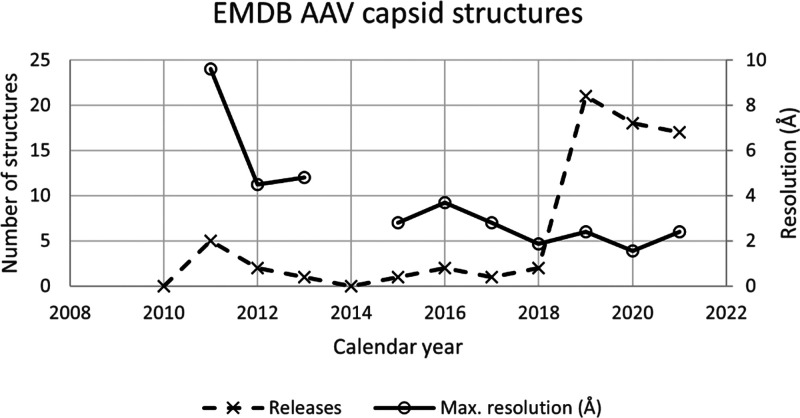
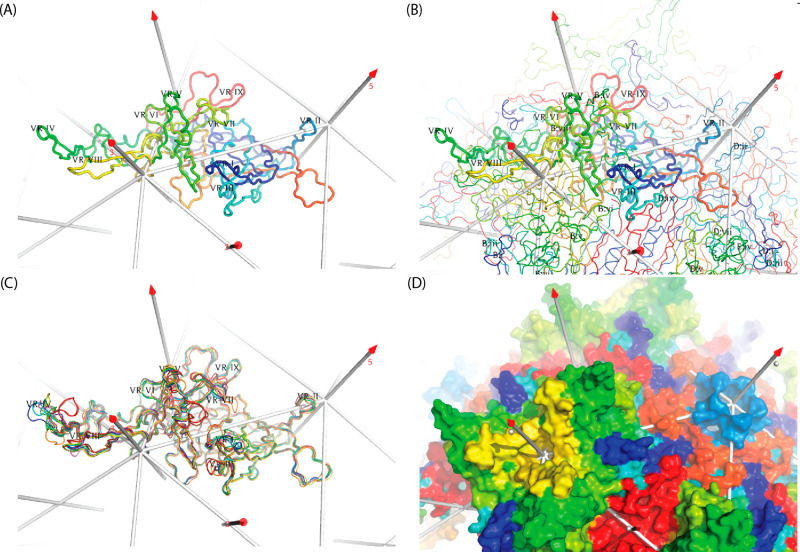
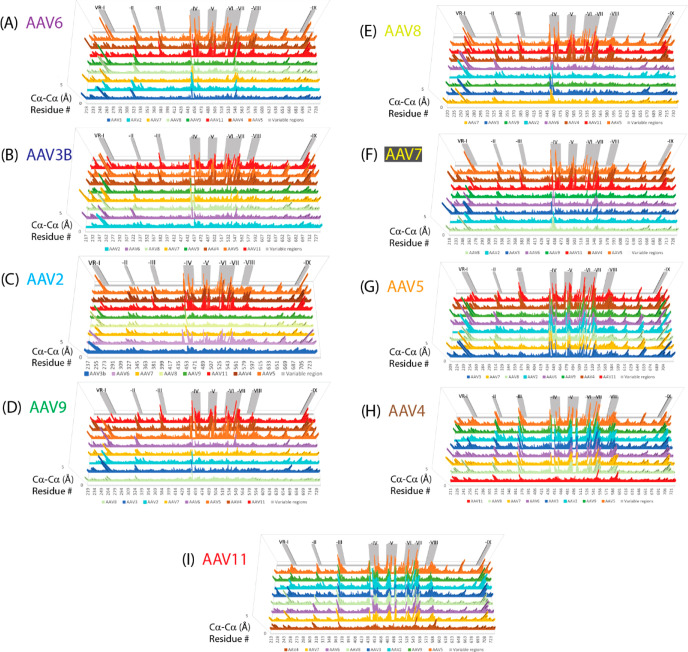
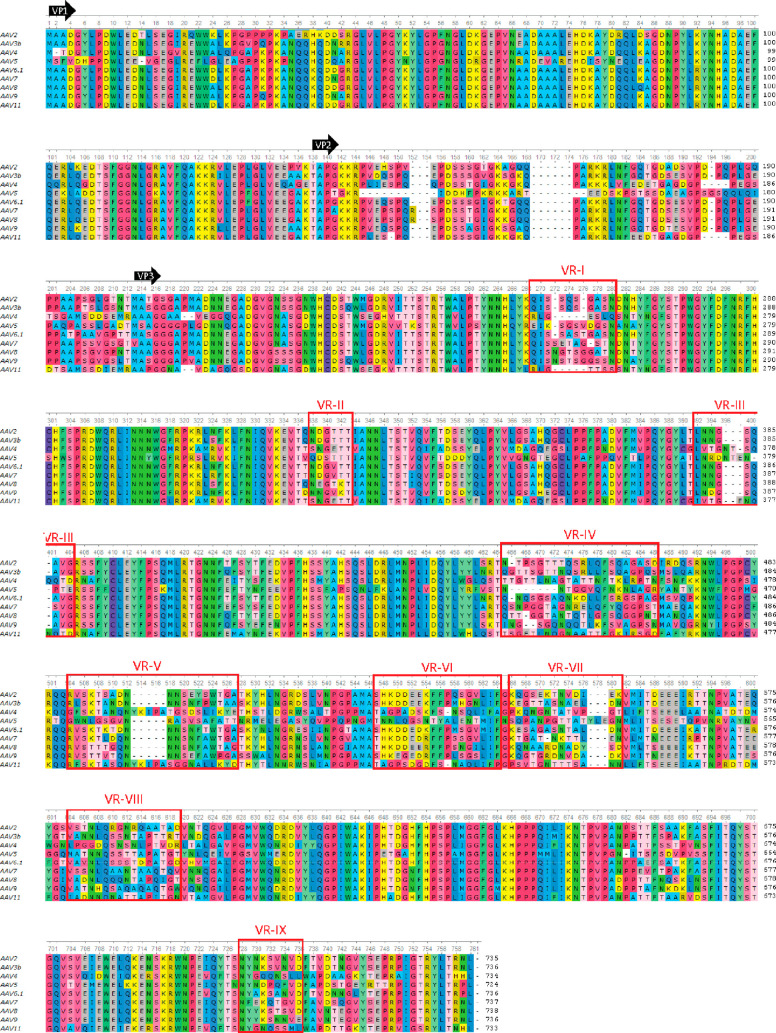
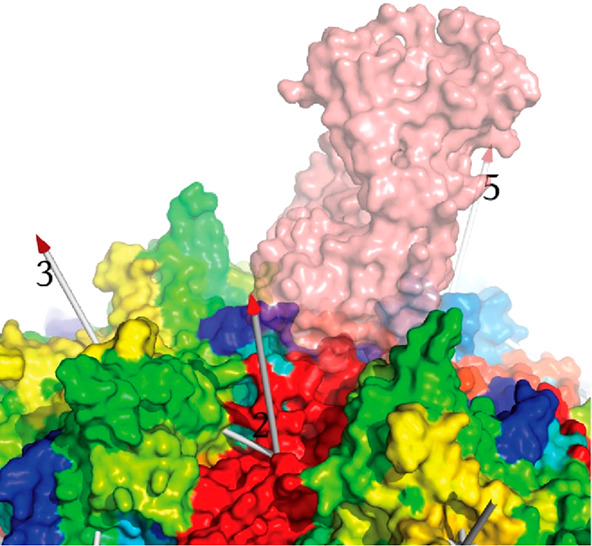
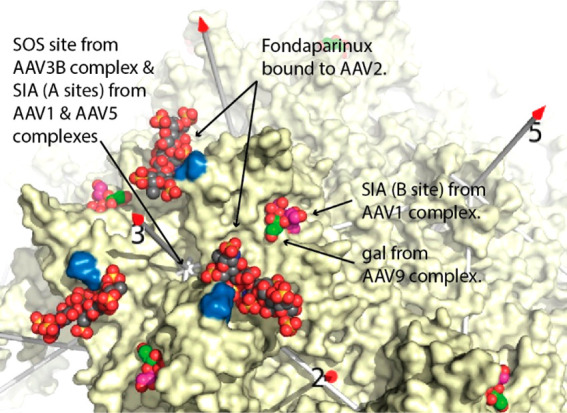
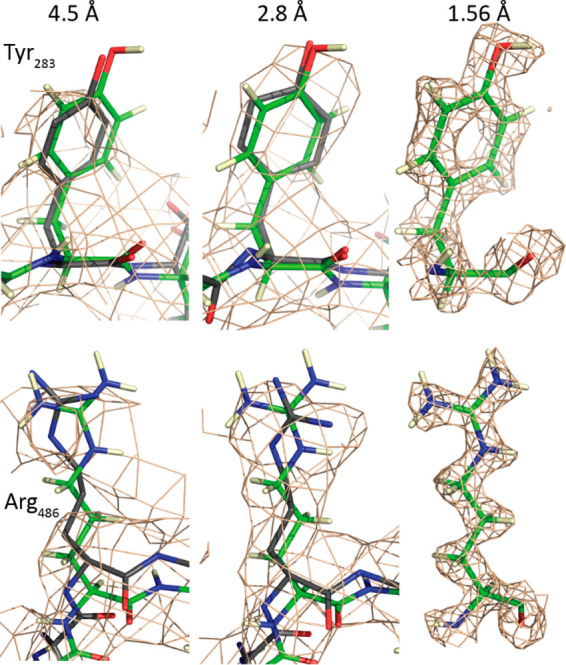
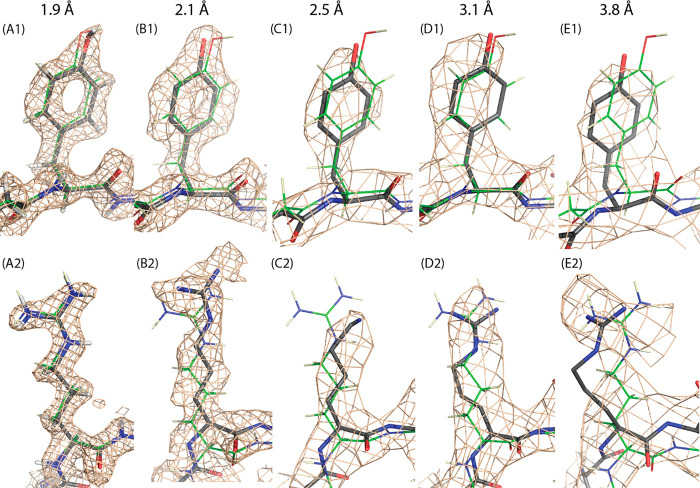
Adeno-associated virus (AAV) has a single-stranded DNA genome encapsidated in a small icosahedrally symmetric protein shell with 60 subunits. AAV is the leading delivery vector in emerging gene therapy treatments for inherited disorders, so its structure and molecular interactions with human hosts are of intense interest. A wide array of electron microscopic approaches have been used to visualize the virus and its complexes, depending on the scientific question, technology available, and amenability of the sample. Approaches range from subvolume tomographic analyses of complexes with large and flexible host proteins to detailed analysis of atomic interactions within the virus and with small ligands at resolutions as high as 1.6 Å. Analyses have led to the reclassification of glycan receptors as attachment factors, to structures with a new-found receptor protein, to identification of the epitopes of antibodies, and a new understanding of possible neutralization mechanisms. AAV is now well-enough characterized that it has also become a model system for EM methods development. Heralding a new era, cryo-EM is now also being deployed as an analytic tool in the process development and production quality control of high value pharmaceutical biologics, namely AAV vectors.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


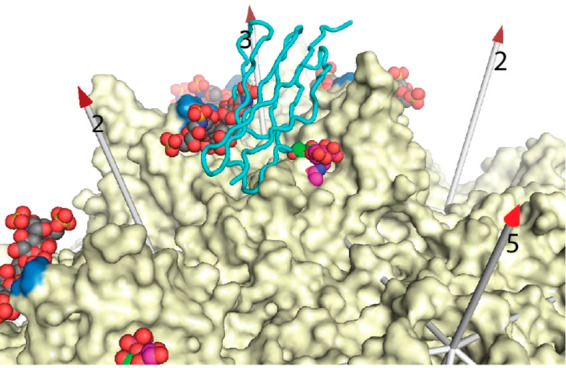
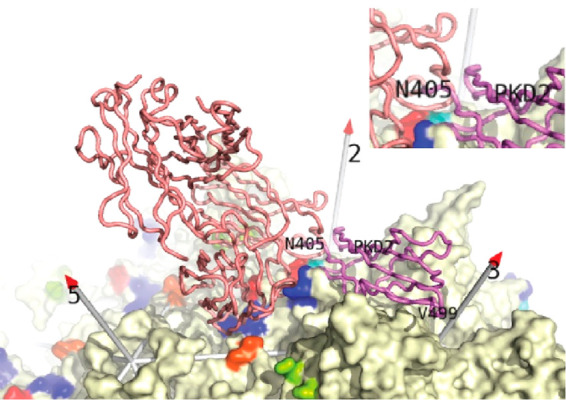
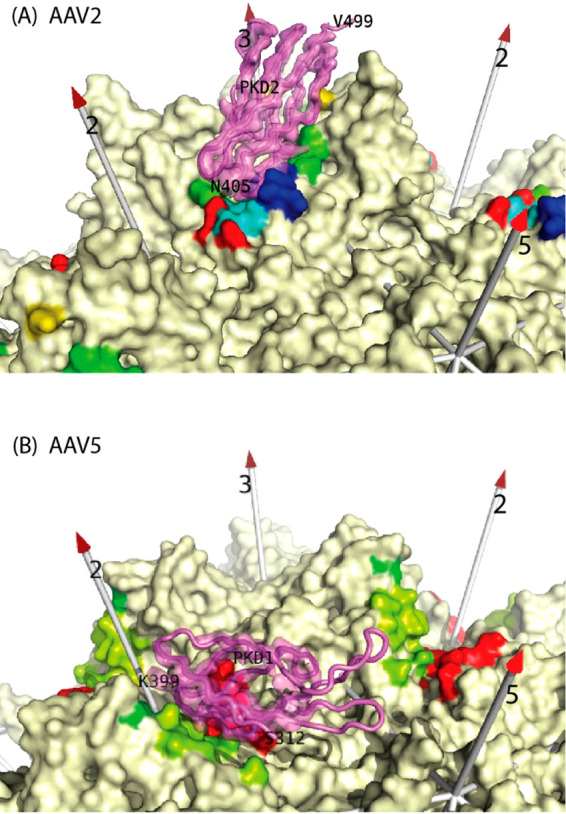
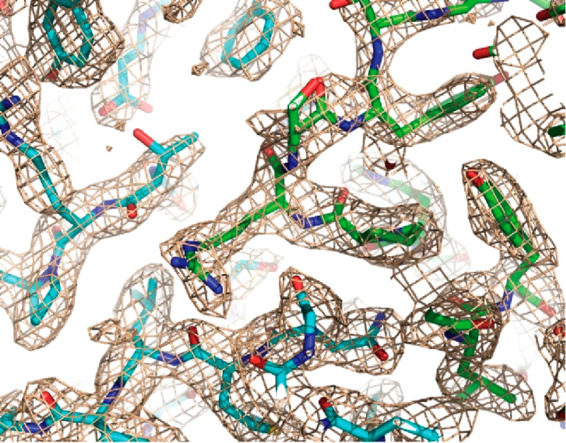
References
-
- Berns K. I., Parvoviridae: The Viruses and Their Replication. In Virology, 3rd ed.; Fields B. N., Knipe D. M., Howley P. M., Eds.; Raven Press: Philadelphia, 1996; pp 1017–1041.
-
- Harrison S. C.Principles of Virus Structure. In Virology, Fields B. N., Knipe D. M., Eds.; Raven Press: New York, 1990; pp 37–61.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
