

Bi-alignments with affine gaps costs
- PMID: 35578255
- PMCID: PMC9109335
- DOI: 10.1186/s13015-022-00219-7
Bi-alignments with affine gaps costs
Abstract
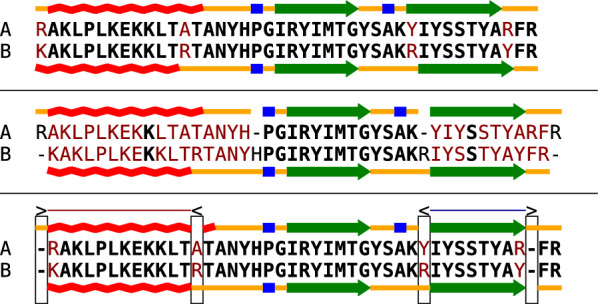
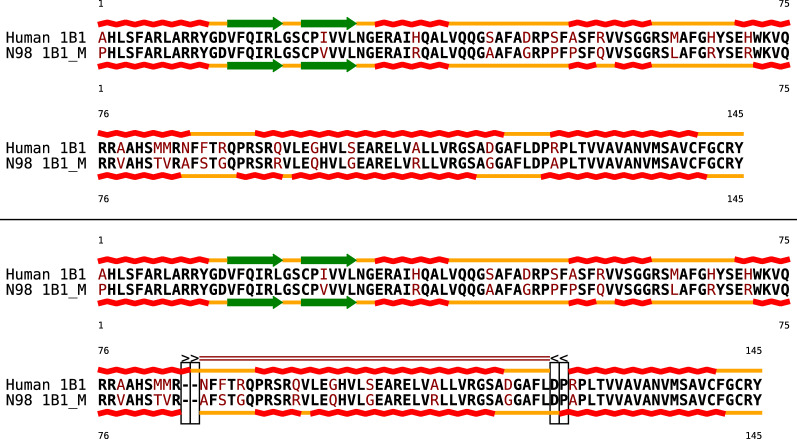
Background: Commonly, sequence and structure elements are assumed to evolve congruently, such that homologous sequence positions correspond to homologous structural features. Assuming congruent evolution, alignments based on sequence and structure similarity can therefore optimize both similarities at the same time in a single alignment. To model incongruent evolution, where sequence and structural features diverge positionally, we recently introduced bi-alignments. This generalization of sequence and structure-based alignments is best understood as alignments of two distinct pairwise alignments of the same entities: one modeling sequence similarity, the other structural similarity.
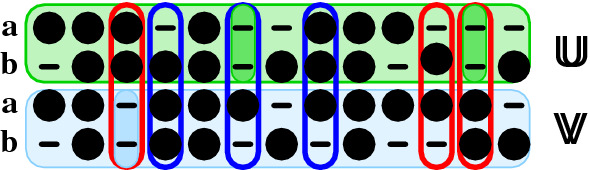
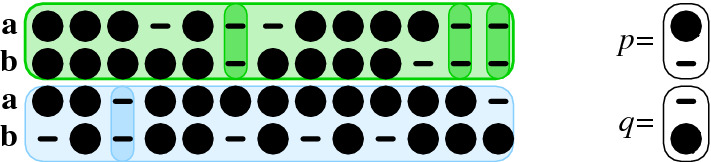
Results: Optimal bi-alignments with affine gap costs (or affine shift cost) for two constituent alignments can be computed exactly in quartic space and time. Even bi-alignments with affine shift and gap cost, as well as bi-alignment with sub-additive gap cost are optimized efficiently. Affine gap-cost bi-alignment of large proteins ([Formula: see text] aa) can be computed.
Conclusion: Affine cost bi-alignments are of practical interest to study shifts of protein sequences and protein structures relative to each other.
Availability: The affine cost bi-alignment algorithm has been implemented in Python 3 and Cython. It is available as free software from https://github.com/s-will/BiAlign/releases/tag/v0.3 and as bioconda package bialign.
Keywords: Dynamic programming; Multi-tape formal grammar; Recursion; Scoring functions.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Wagner GP. Homology, genes, and evolutionary innovation. Princeton: Princeton Univ. Press; 2014.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
