

Feasibility and clinical utility of comprehensive genomic profiling of hematological malignancies
- PMID: 35579198
- PMCID: PMC9357666
- DOI: 10.1111/cas.15427
Feasibility and clinical utility of comprehensive genomic profiling of hematological malignancies
Abstract
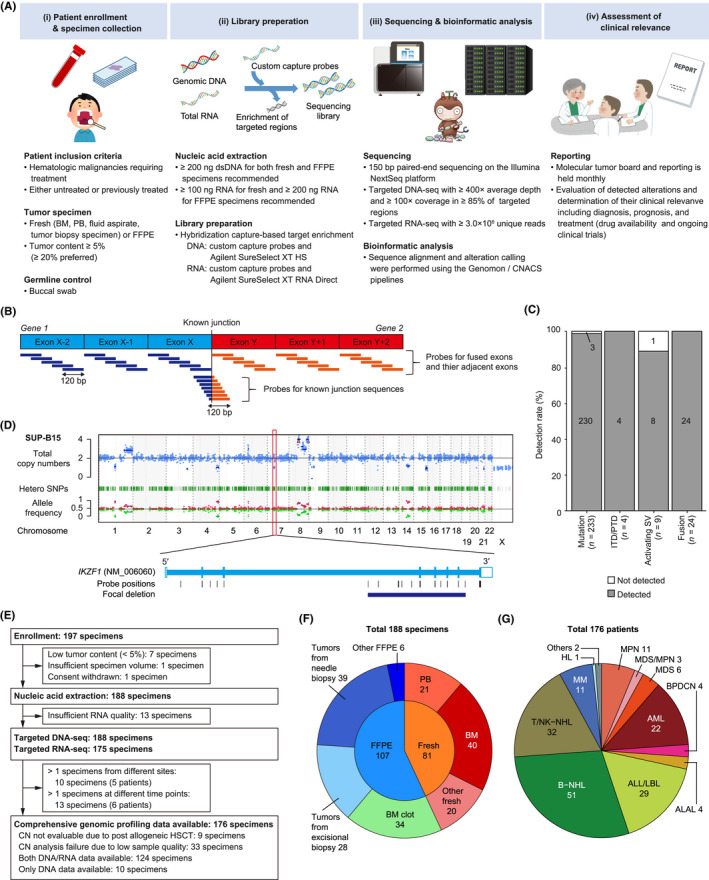
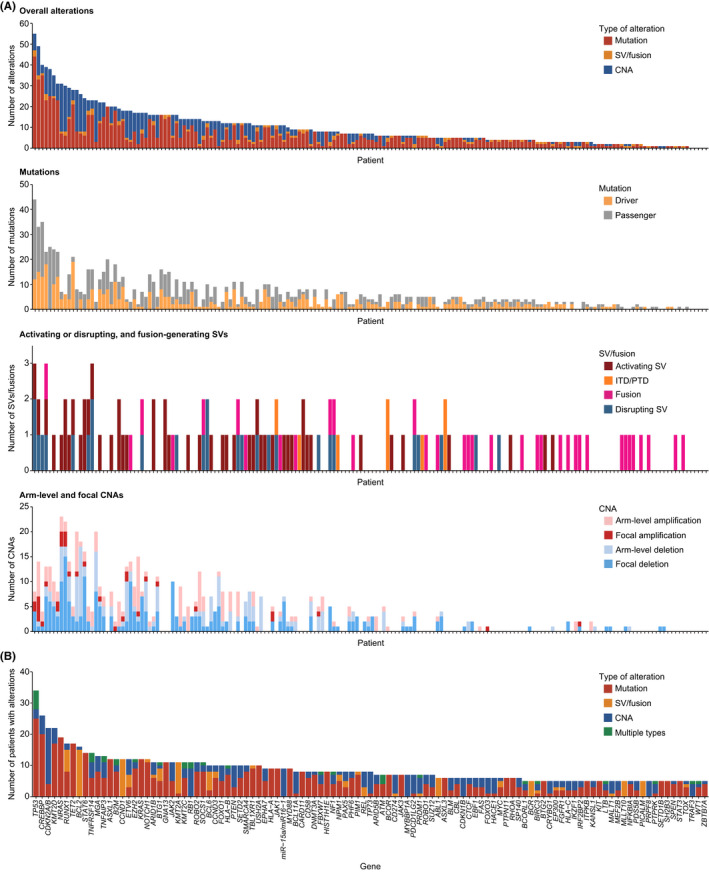
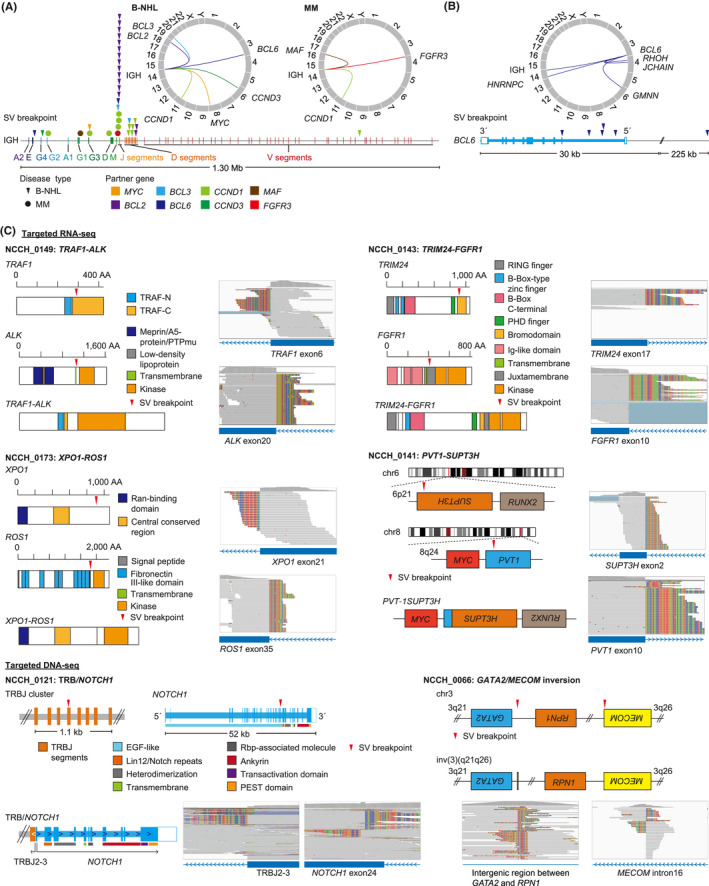
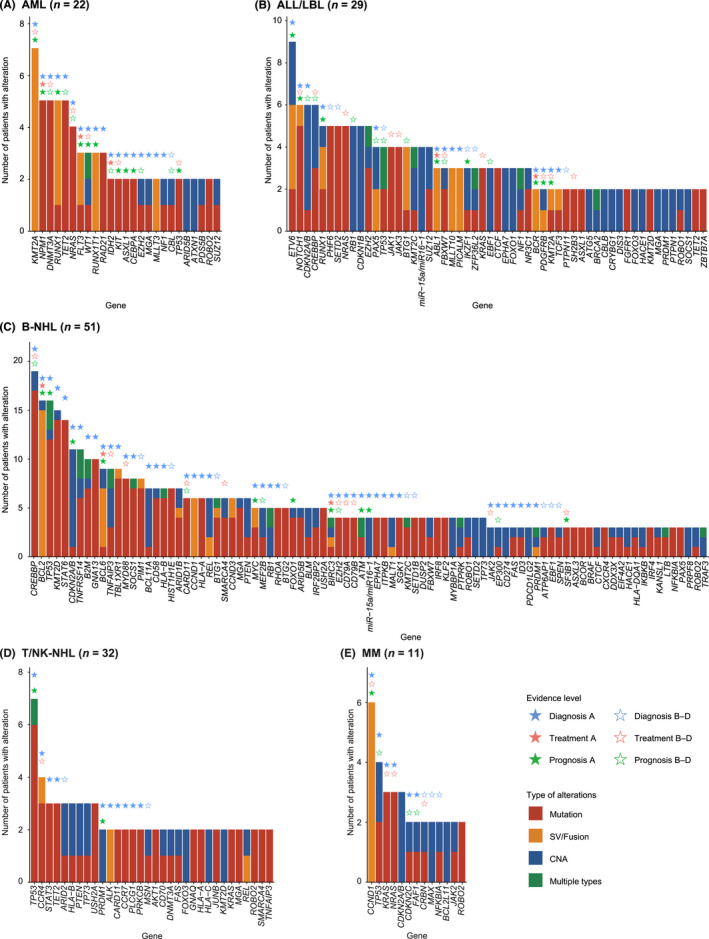
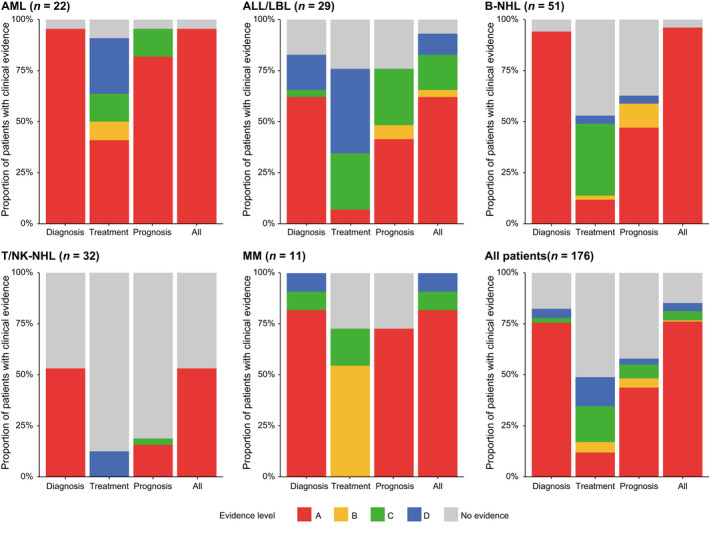
Identification of genetic alterations through next-generation sequencing (NGS) can guide treatment decision-making by providing information on diagnosis, therapy selection, and prognostic stratification in patients with hematological malignancies. Although the utility of NGS-based genomic profiling assays was investigated in hematological malignancies, no assays sufficiently cover driver mutations, including recently discovered ones, as well as fusions and/or pathogenic germline variants. To address these issues, here we have devised an integrated DNA/RNA profiling assay to detect various types of somatic alterations and germline variants at once. Particularly, our assay can successfully identify copy number alterations and structural variations, including immunoglobulin heavy chain translocations, IKZF1 intragenic deletions, and rare fusions. Using this assay, we conducted a prospective study to investigate the feasibility and clinical usefulness of comprehensive genomic profiling for 452 recurrently altered genes in hematological malignancies. In total, 176 patients (with 188 specimens) were analyzed, in which at least one alteration was detected in 171 (97%) patients, with a median number of total alterations of 7 (0-55). Among them, 145 (82%), 86 (49%), and 102 (58%) patients harbored at least one clinically relevant alteration for diagnosis, treatment, and prognosis, respectively. The proportion of patients with clinically relevant alterations was the highest in acute myeloid leukemia, whereas this assay was less informative in T/natural killer-cell lymphoma. These results suggest the clinical utility of NGS-based genomic profiling, particularly for their diagnosis and prognostic prediction, thereby highlighting the promise of precision medicine in hematological malignancies.
Keywords: comprehensive genomic profiling; hematological malignancy; next-generation sequencing; precision medicine; somatic alteration.
© 2022 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Figures
References
-
- McDermott U. Next‐generation sequencing and empowering personalised cancer medicine. Drug Discov Today. 2015;20:1470‐1475. - PubMed
-
- Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th Edition ed. International Agency for Research on Cancer; 2017.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
