

Modulation of IKs channel-PIP2 interaction by PRMT1 plays a critical role in the control of cardiac repolarization
- PMID: 35580065
- PMCID: PMC9543859
- DOI: 10.1002/jcp.30775
Modulation of IKs channel-PIP2 interaction by PRMT1 plays a critical role in the control of cardiac repolarization
Abstract
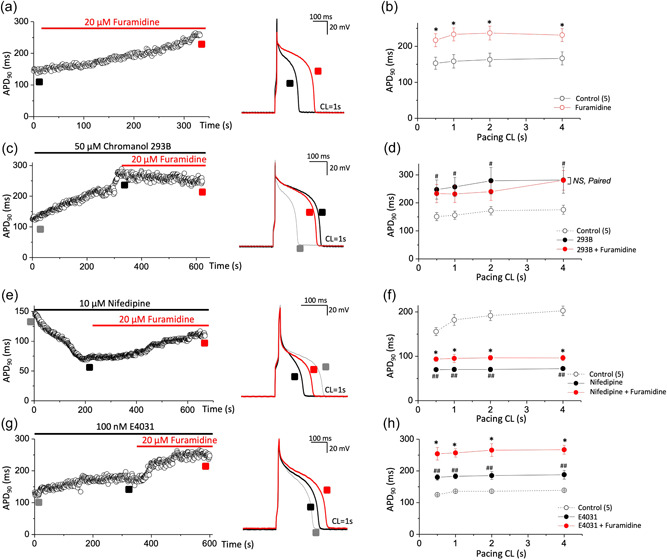
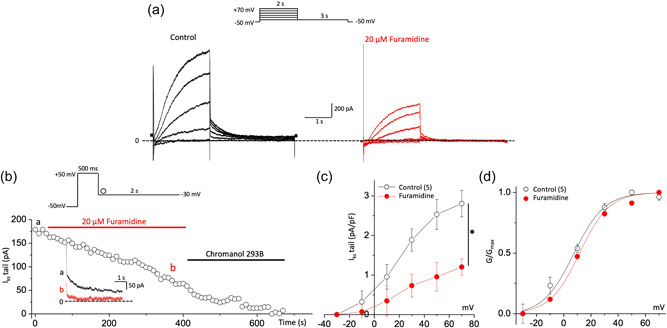
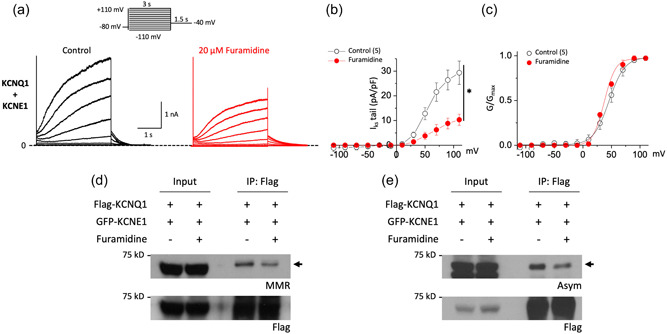
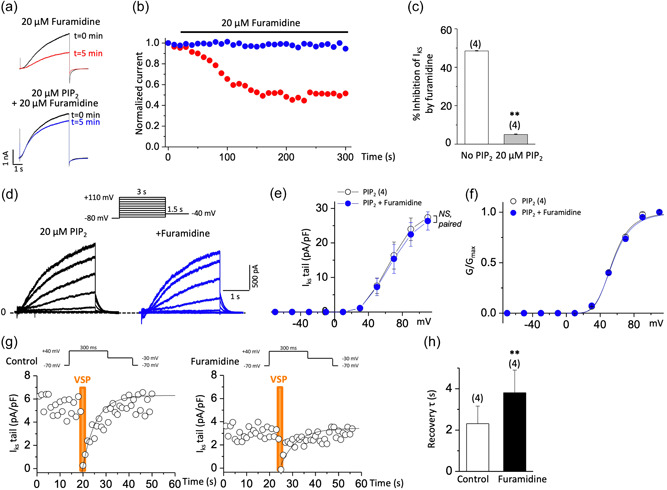
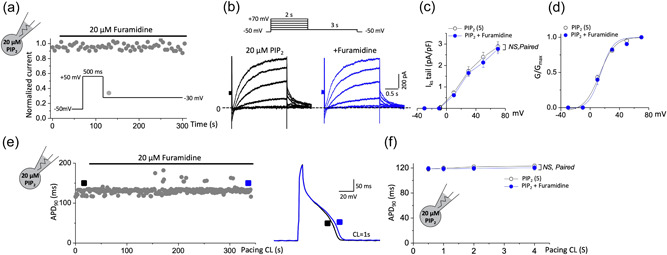
Recent studies have shown that protein arginine methyltransferase 1 (PRMT1) is highly expressed in the human heart, and loss of PRMT1 contributes to cardiac remodeling in the heart failure. However, the functional importance of PRMT1 in cardiac ion channels remains uncertain. The slow activating delayed rectifier K+ (IKs ) channel is a cardiac K+ channel composed of KCNQ1 and KCNE1 subunits and is a new therapeutic target for treating lethal arrhythmias in many cardiac pathologies, especially heart failure. Here, we demonstrate that PRMT1 is a critical regulator of the IKs channel and cardiac rhythm. In the guinea pig ventricular myocytes, treatment with furamidine, a PRMT1-specific inhibitor, prolonged the action potential duration (APD). We further show that this APD prolongation was attributable to IKs reduction. In HEK293T cells expressing human KCNQ1 and KCNE1, inhibiting PRMT1 via furamidine reduced IKs and concurrently decreased the arginine methylation of KCNQ1, a pore-forming α-subunit. Evidence presented here indicates that furamidine decreased IKs mainly by lowering the affinity of IKs channels for the membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2 ), which is crucial for pore opening. Finally, applying exogenous PIP2 to cardiomyocytes prevented the furamidine-induced IKs reduction and APD prolongation. Taken together, these results indicate that PRMT1 positively regulated IKs activity through channel-PIP2 interaction, thereby restricting excessive cardiac action potential.
Keywords: PIP2; PRMT1; arrhythmia; cardiac myocytes; delayed rectifier potassium channel.
© 2022 The Authors. Journal of Cellular Physiology published by Wiley Periodicals LLC.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Aflaki, M. , Qi, X. Y. , Xiao, L. , Ordog, B. , Tadevosyan, A. , Luo, X. , Maguy, A. , Shi, Y. , Tardif, J. C. , & Nattel, S. (2014). Exchange protein directly activated by cAMP mediates slow delayed‐rectifier current remodeling by sustained β‐adrenergic activation in guinea pig hearts. Circulation Research, 114(6), 993–1003. 10.1161/circresaha.113.302982 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
