

DNA methylation clocks for dogs and humans
- PMID: 35580182
- PMCID: PMC9173771
- DOI: 10.1073/pnas.2120887119
DNA methylation clocks for dogs and humans
Abstract
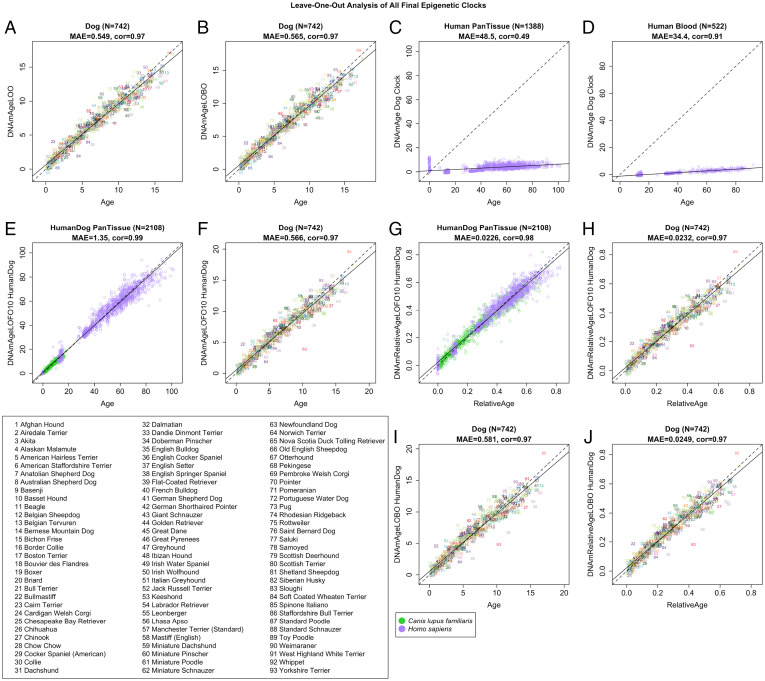
DNA methylation profiles have been used to develop biomarkers of aging known as epigenetic clocks, which predict chronological age with remarkable accuracy and show promise for inferring health status as an indicator of biological age. Epigenetic clocks were first built to monitor human aging, but their underlying principles appear to be evolutionarily conserved, as they have now been successfully developed for many mammalian species. Here, we describe reliable and highly accurate epigenetic clocks shown to apply to 93 domestic dog breeds. The methylation profiles were generated using the mammalian methylation array, which utilizes DNA sequences that are conserved across all mammalian species. Canine epigenetic clocks were constructed to estimate age and also average time to death. We also present two highly accurate human–dog dual species epigenetic clocks (R = 0.97), which may facilitate the ready translation from canine to human use (or vice versa) of antiaging treatments being developed for longevity and preventive medicine. Finally, epigenome-wide association studies here reveal individual methylation sites that may underlie the inverse relationship between breed weight and lifespan. Overall, we describe robust biomarkers to measure aging and, potentially, health status in canines.
Keywords: Canis familiaris; aging; dog; epigenetic clock; methylation.
Conflict of interest statement
Competing interest statement: S.H. is a founder of the nonprofit Epigenetic Clock Development Foundation who plans to license several patents from his employer University of California (UC) Regents. These patents list S.H. as an inventor. R.T.B. is the Executive Director of the Epigenetic Clock Development Foundation.
Figures





References
-
- Gilmore K. M., Greer K. A., Why is the dog an ideal model for aging research? Exp. Gerontol. 71, 14–20 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
