

Myotubularin-related phosphatase 5 is a critical determinant of autophagy in neurons
- PMID: 35580604
- PMCID: PMC9233098
- DOI: 10.1016/j.cub.2022.04.053
Myotubularin-related phosphatase 5 is a critical determinant of autophagy in neurons
Abstract
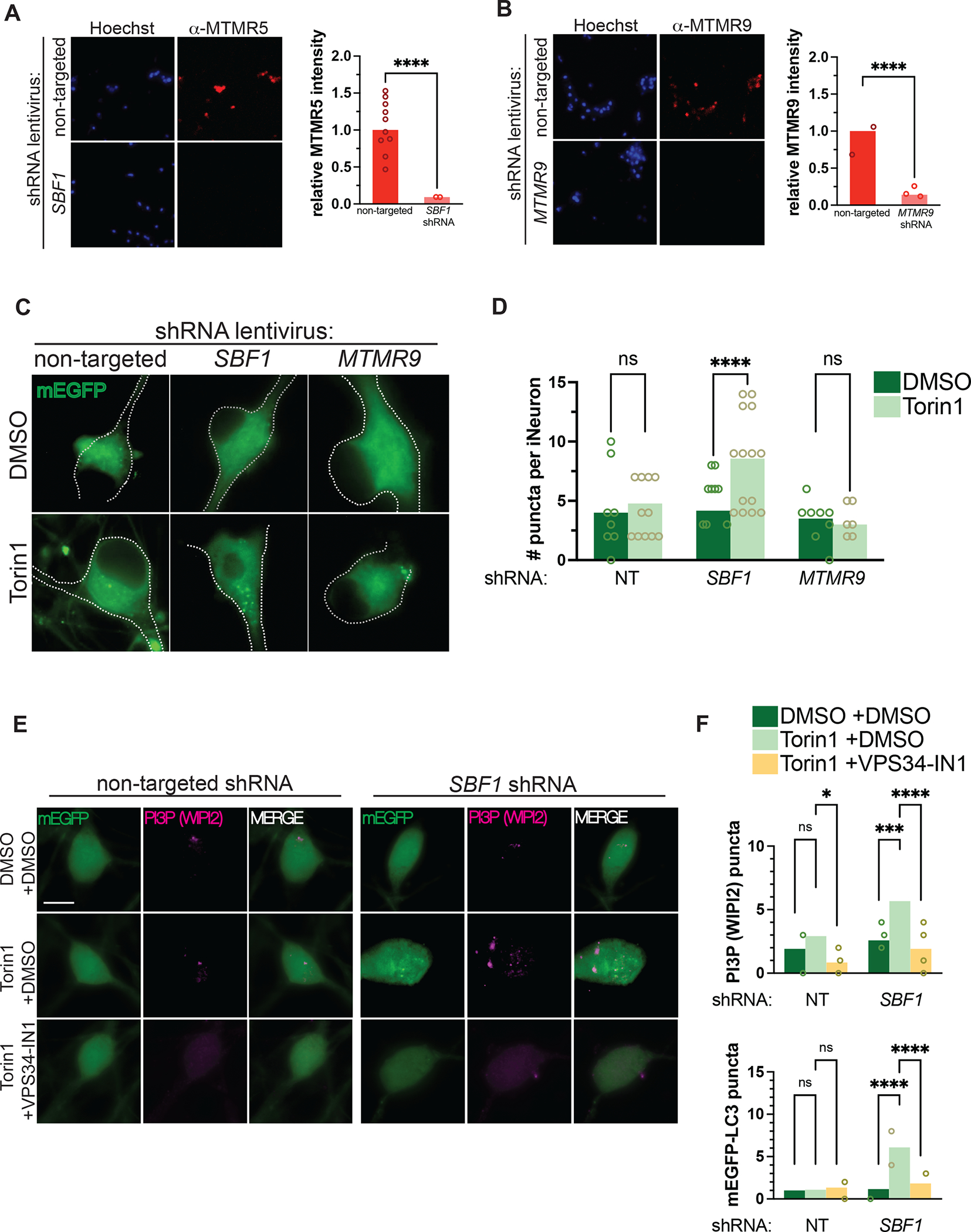
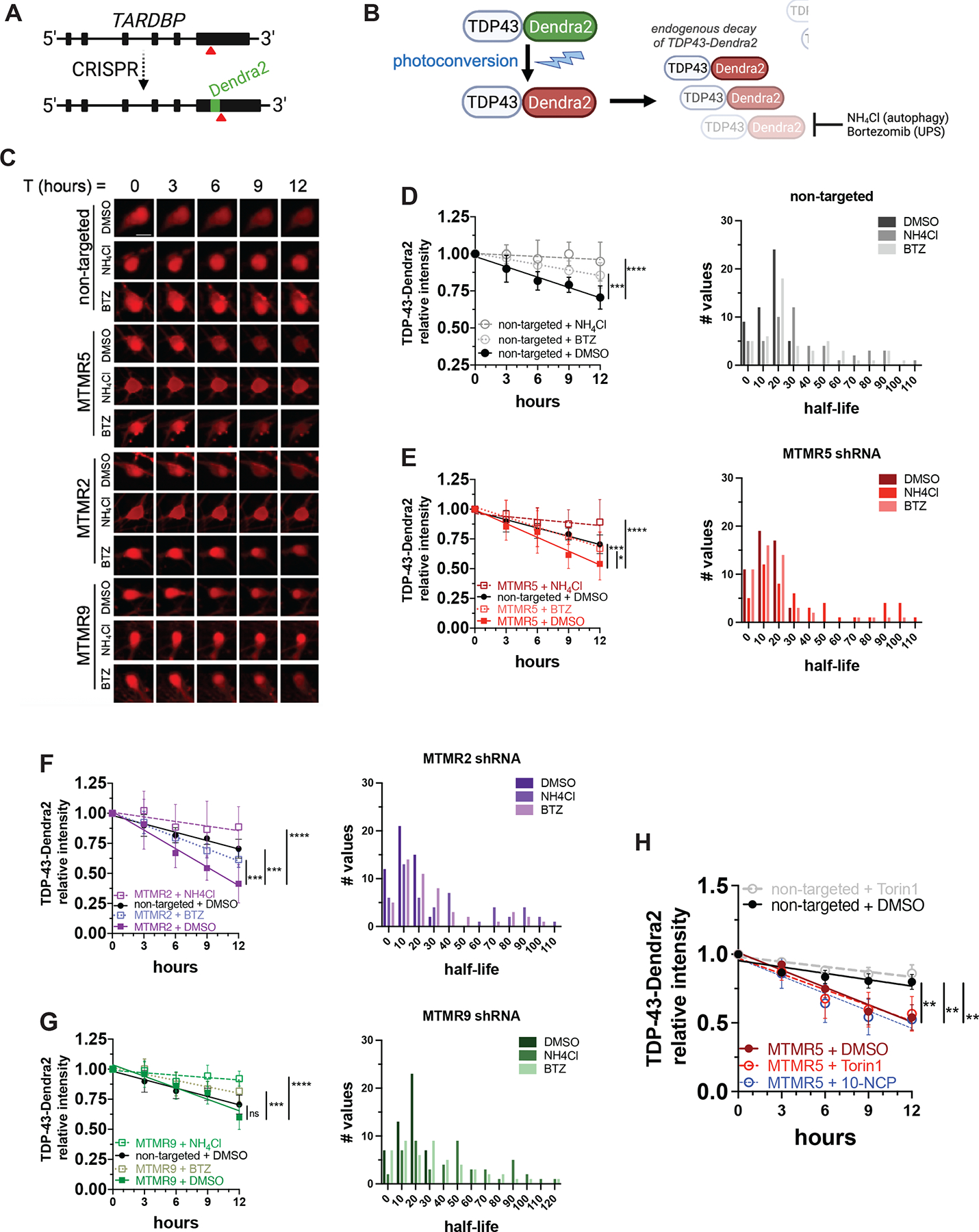
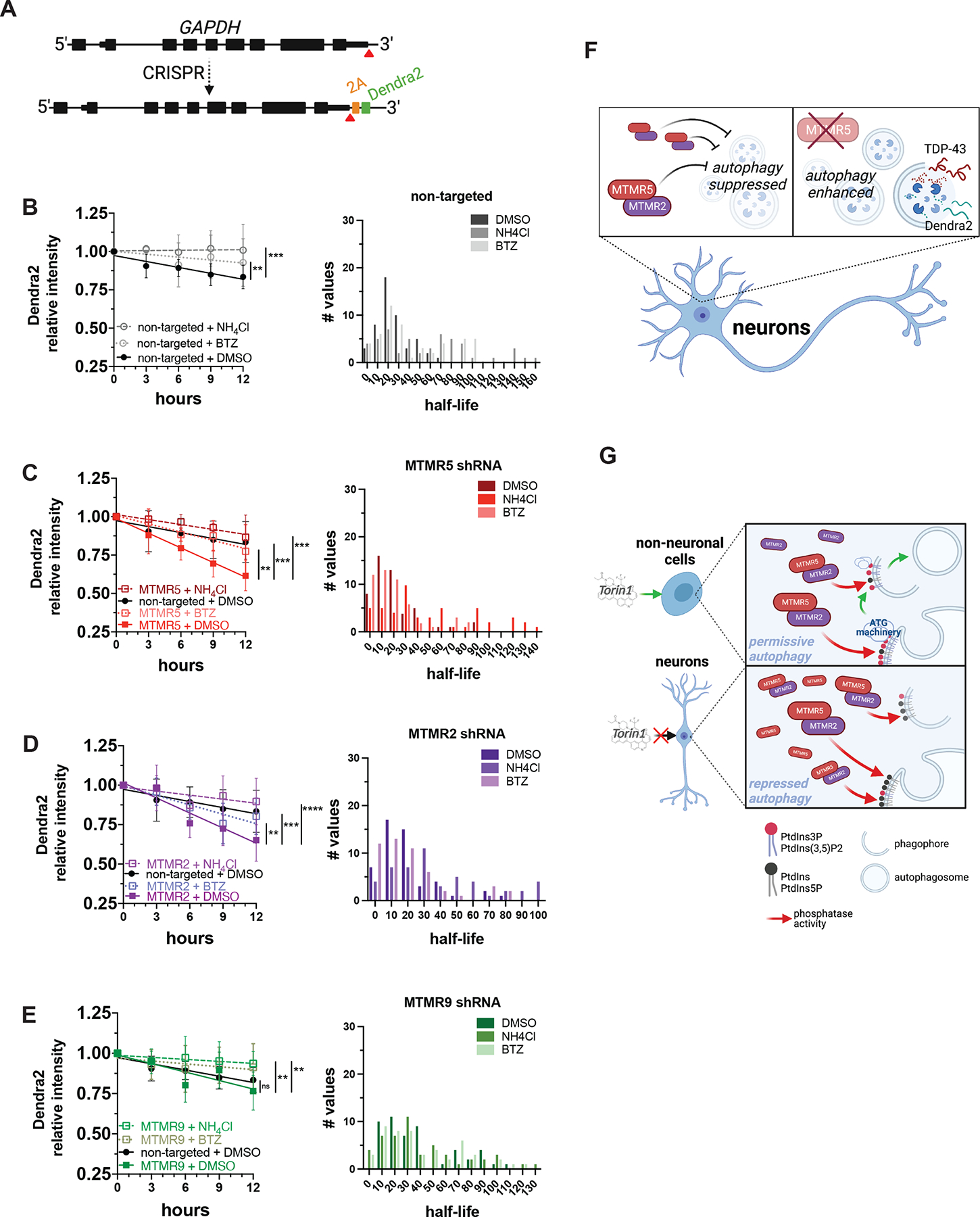
Autophagy is a conserved, multi-step process of capturing proteolytic cargo in autophagosomes for lysosome degradation. The capacity to remove toxic proteins that accumulate in neurodegenerative disorders attests to the disease-modifying potential of the autophagy pathway. However, neurons respond only marginally to conventional methods for inducing autophagy, limiting efforts to develop therapeutic autophagy modulators for neurodegenerative diseases. The determinants underlying poor autophagy induction in neurons and the degree to which neurons and other cell types are differentially sensitive to autophagy stimuli are incompletely defined. Accordingly, we sampled nascent transcript synthesis and stabilities in fibroblasts, induced pluripotent stem cells (iPSCs), and iPSC-derived neurons (iNeurons), thereby uncovering a neuron-specific stability of transcripts encoding myotubularin-related phosphatase 5 (MTMR5). MTMR5 is an autophagy suppressor that acts with its binding partner, MTMR2, to dephosphorylate phosphoinositides critical for autophagy initiation and autophagosome maturation. We found that MTMR5 is necessary and sufficient to suppress autophagy in iNeurons and undifferentiated iPSCs. Using optical pulse labeling to visualize the turnover of endogenously encoded proteins in live cells, we observed that knockdown of MTMR5 or MTMR2, but not the unrelated phosphatase MTMR9, significantly enhances neuronal degradation of TDP-43, an autophagy substrate implicated in several neurodegenerative diseases. Our findings thus establish a regulatory mechanism of autophagy intrinsic to neurons and targetable for clearing disease-related proteins in a cell-type-specific manner. In so doing, our results not only unravel novel aspects of neuronal biology and proteostasis but also elucidate a strategy for modulating neuronal autophagy that could be of high therapeutic potential for multiple neurodegenerative diseases.
Keywords: RNA stability; TDP-43; autophagosome; iPSCs; induced pluripotent stem cells; macroautophagy; myotubularin; neuronal autophagy; optical pulse labeling; phosphoinositide.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
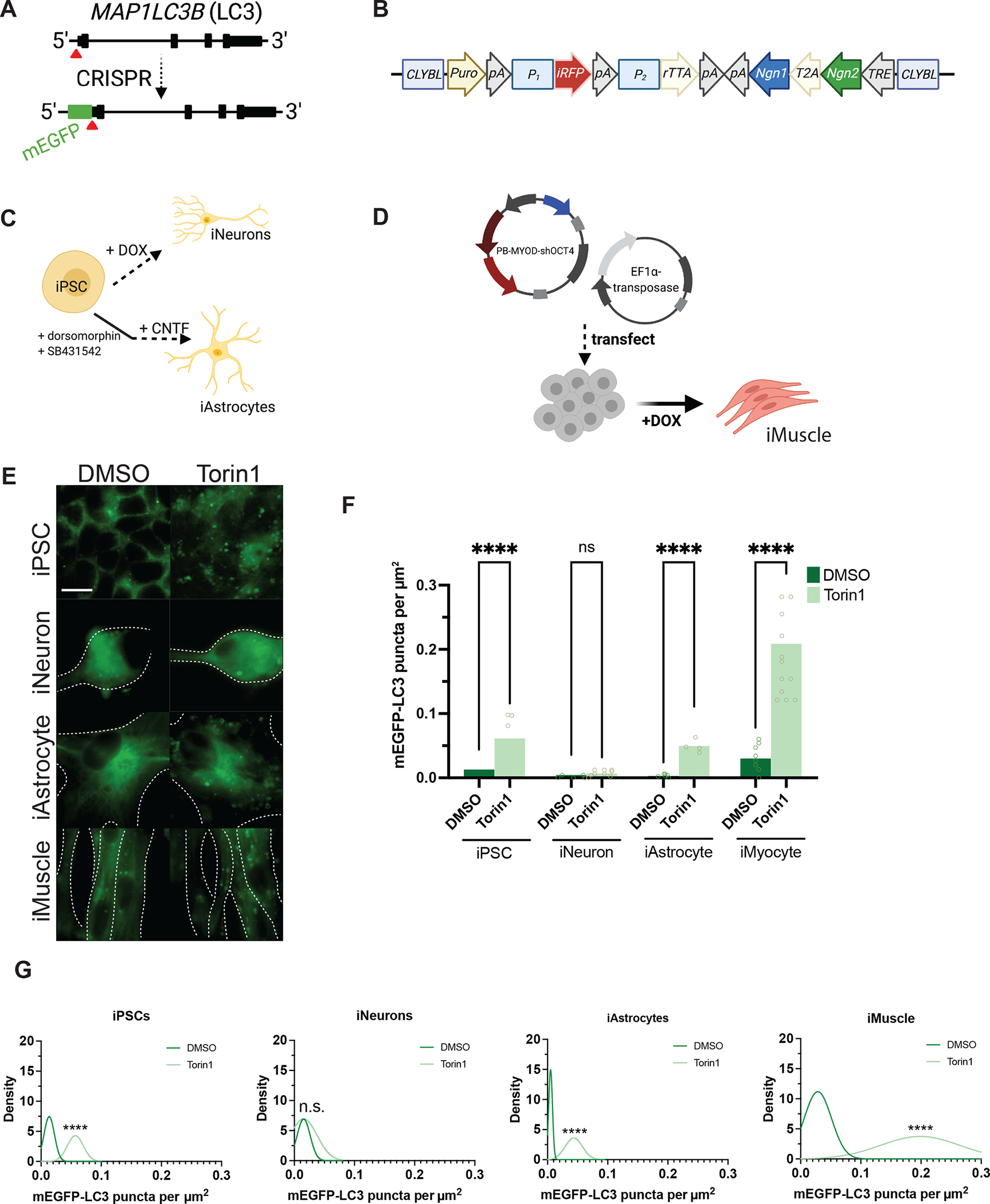
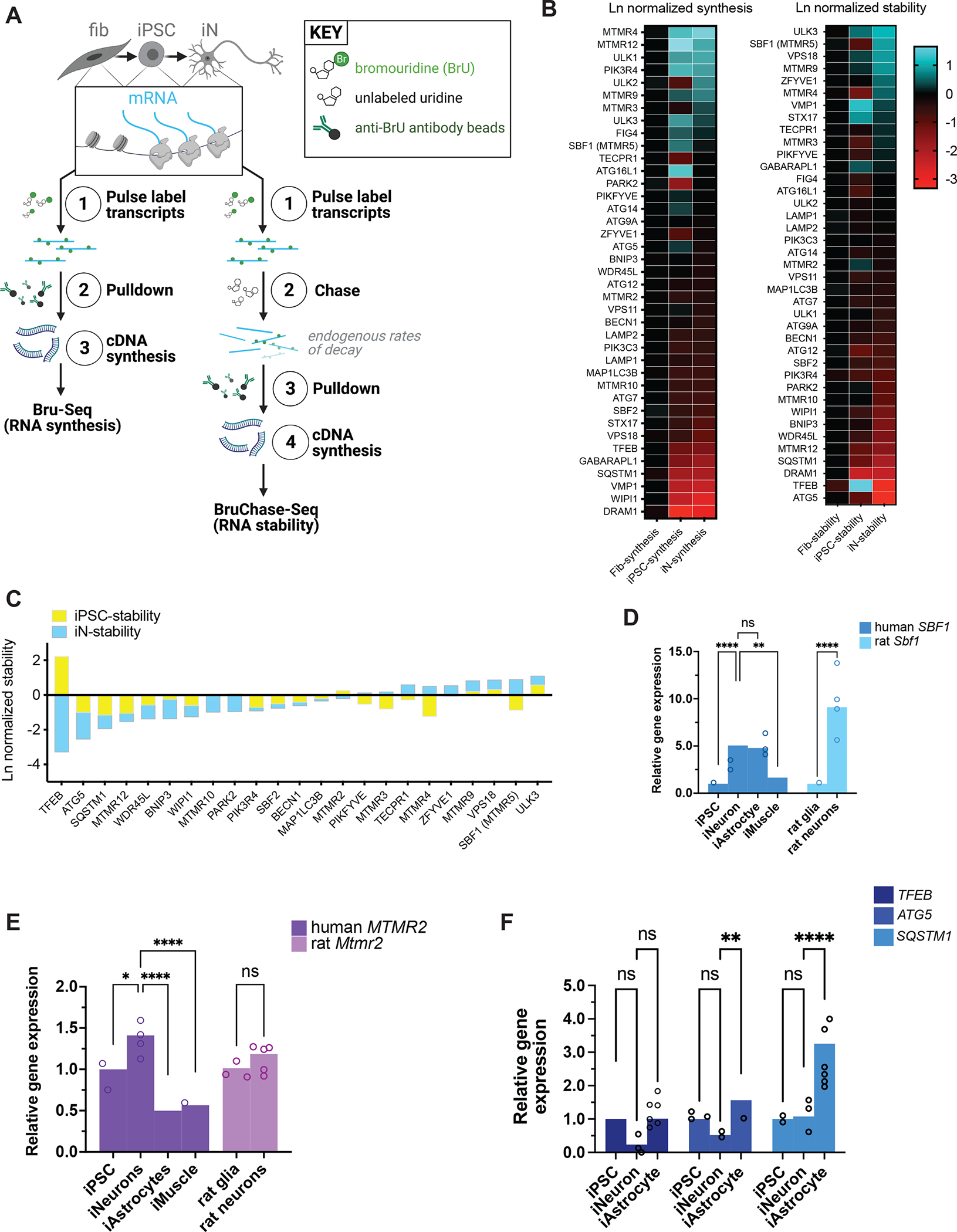
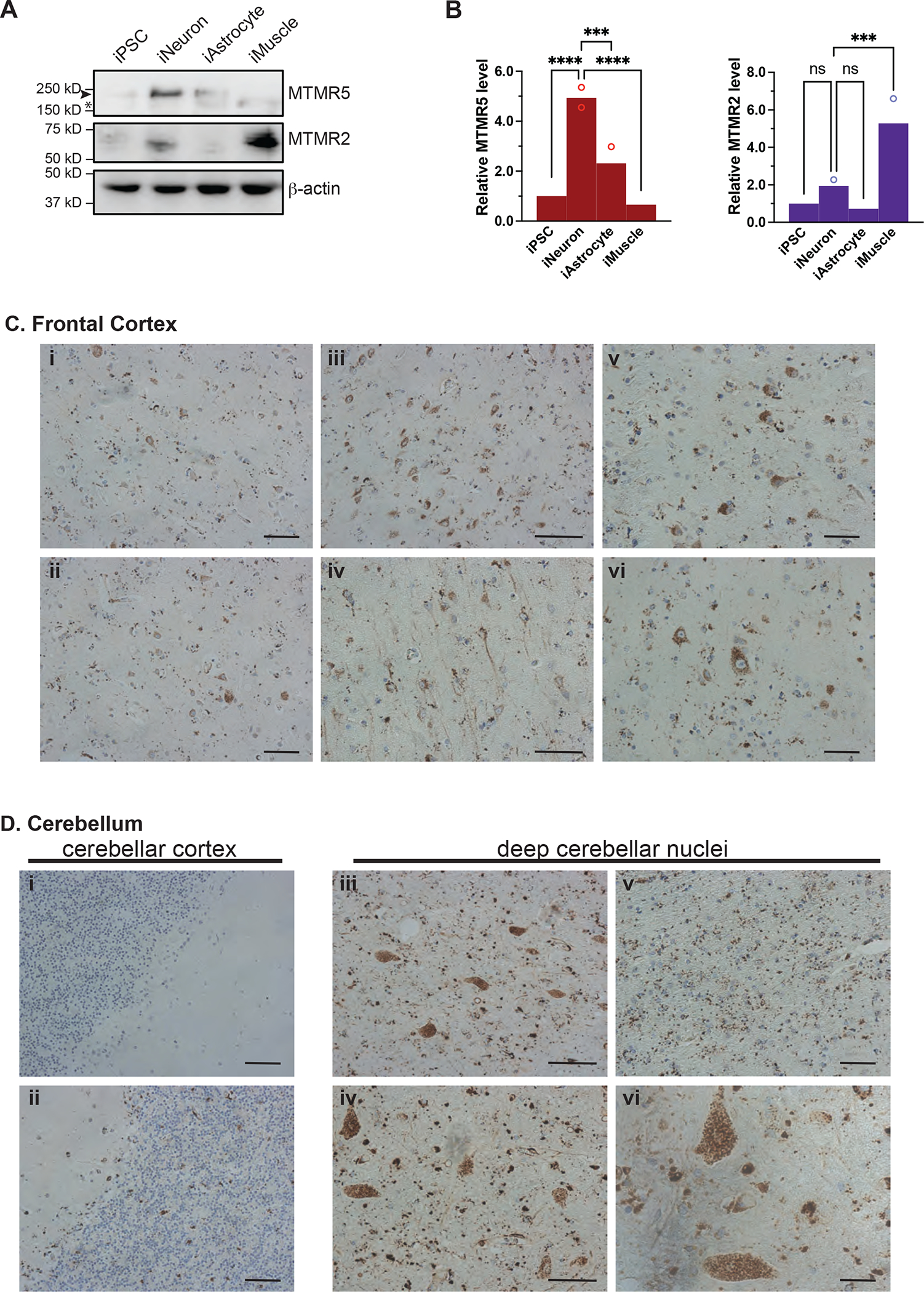
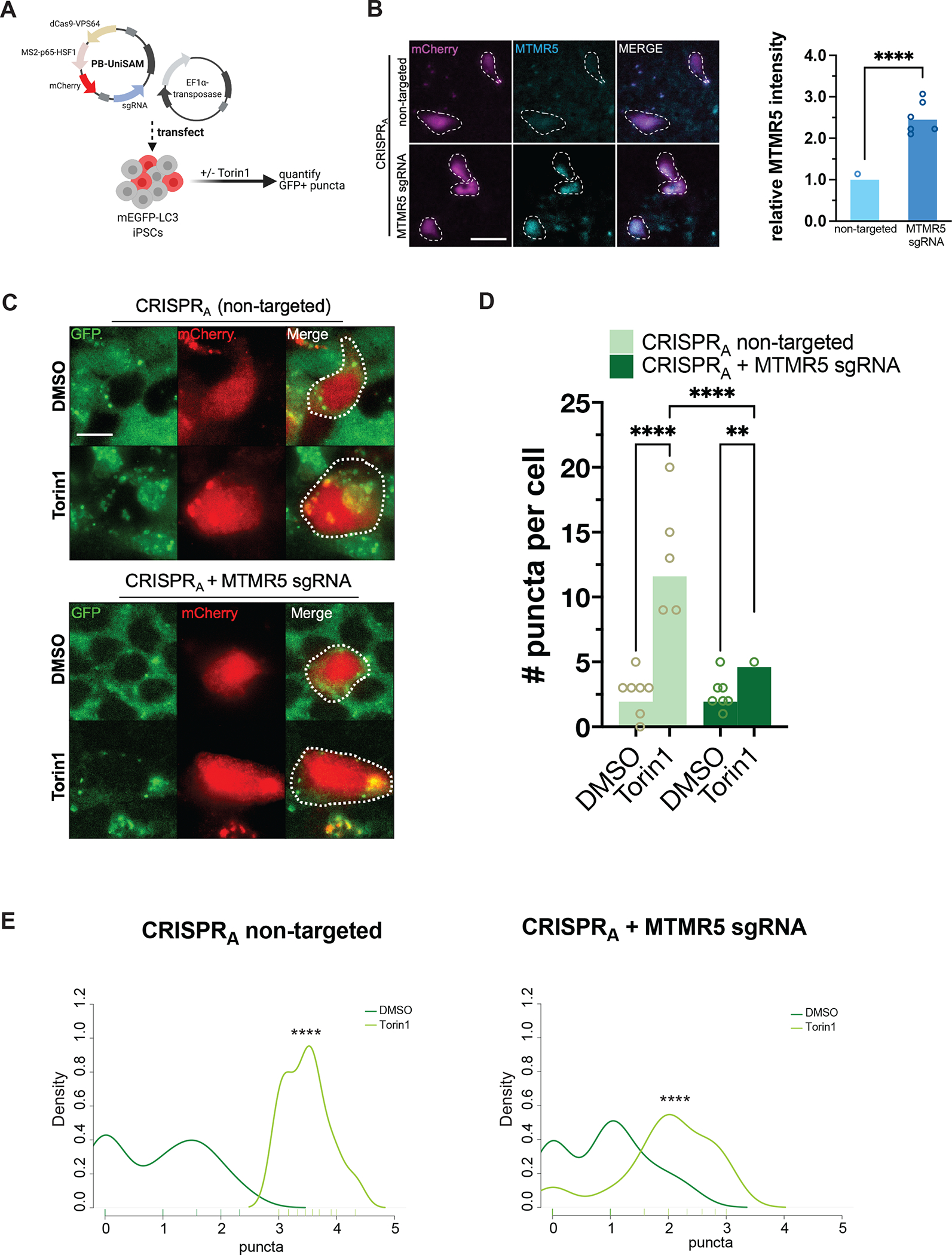
Comment in
-
Autophagy: Identification of MTMR5 as a neuron-enriched suppressor.Curr Biol. 2022 Jun 20;32(12):R574-R577. doi: 10.1016/j.cub.2022.04.075. Curr Biol. 2022. PMID: 35728530 Free PMC article.
References
-
- Claude-Taupin A, and Morel E (2021). Phosphoinositides: Functions in autophagy-related stress responses. Biochim Biophys Acta Mol Cell Biol Lipids 1866, 158903. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
