

Colony‑stimulating factor CSF2 mediates the phenotypic plasticity of small‑cell lung cancer by regulating the p‑STAT3/MYC pathway
- PMID: 35583004
- PMCID: PMC9164265
- DOI: 10.3892/or.2022.8333
Colony‑stimulating factor CSF2 mediates the phenotypic plasticity of small‑cell lung cancer by regulating the p‑STAT3/MYC pathway
Abstract
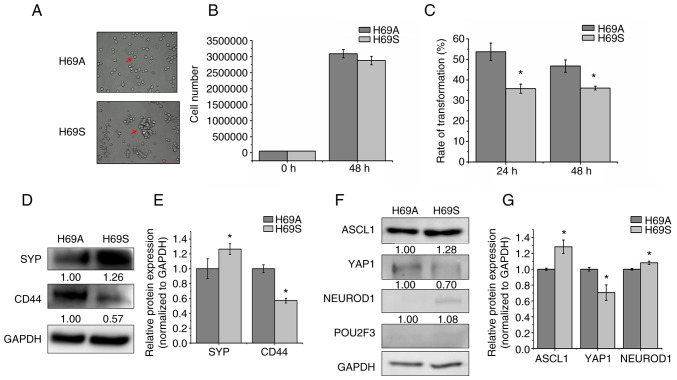
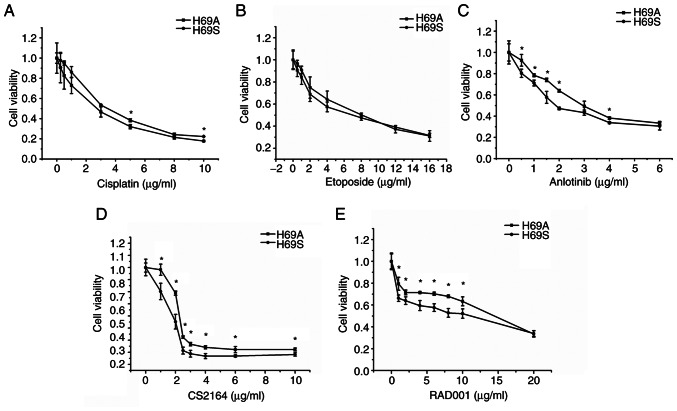
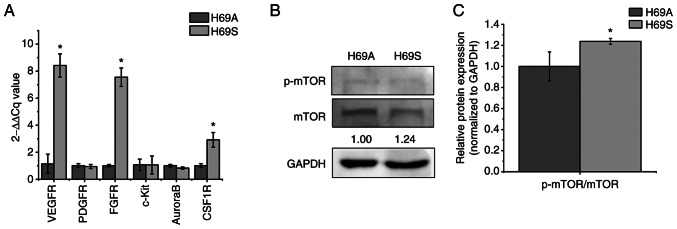
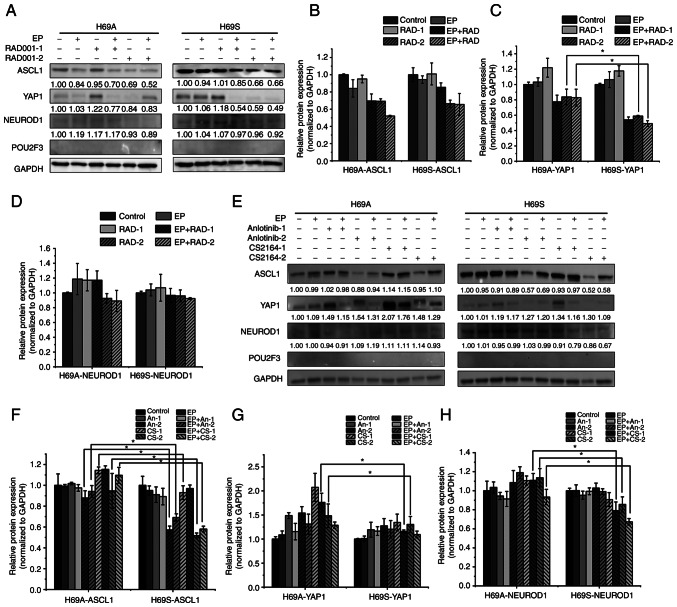
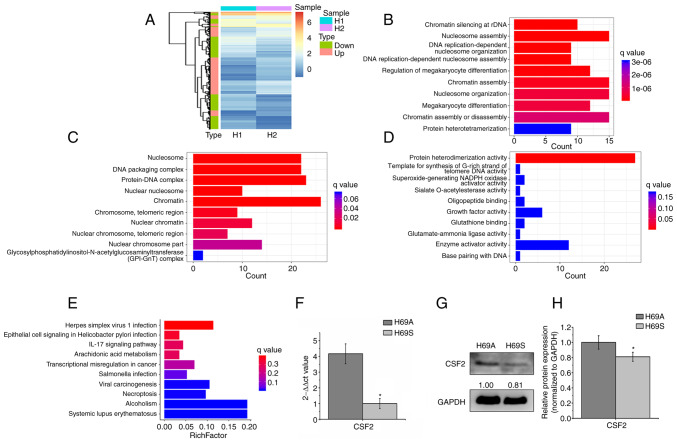
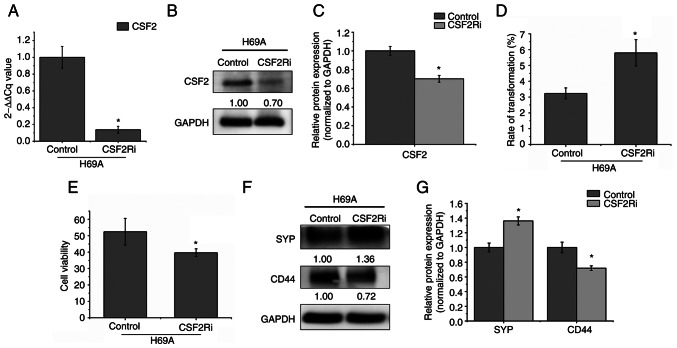
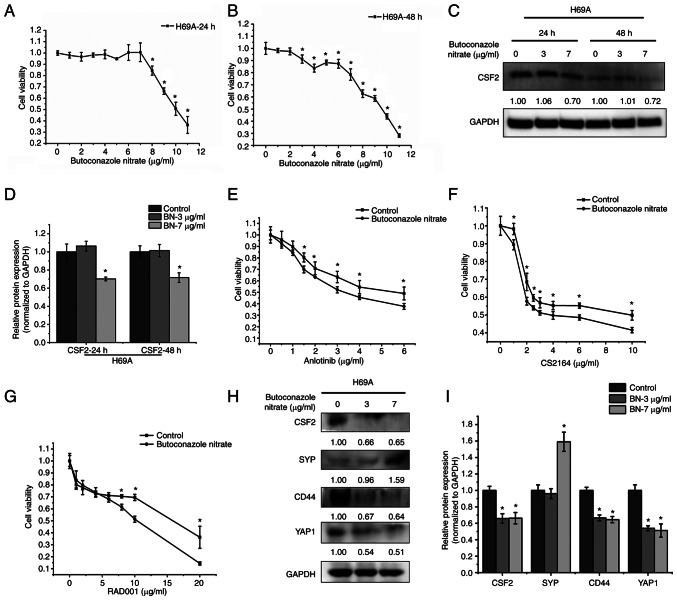
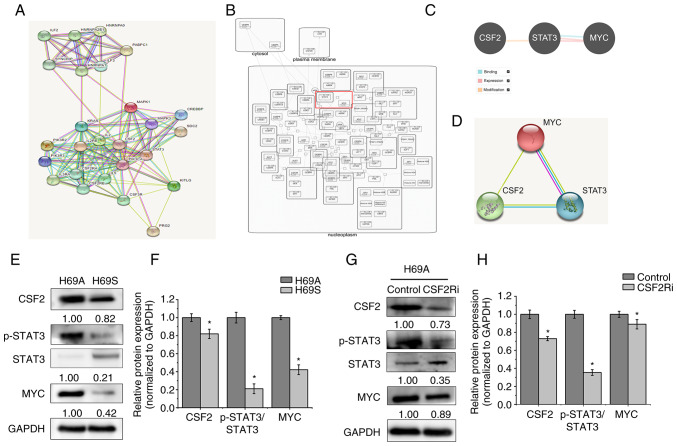
Relapse and drug resistance are the main causes of mortality in patients with small‑cell lung cancer (SCLC). Intratumoral heterogeneity (ITH) is a key biological mechanism that leads to relapse and drug resistance. Phenotypic plasticity is an important factor that leads to ITH in SCLC, although its mechanisms and key regulatory factors remain to be elucidated. In the present study, cell proliferation and cell switch assay were measured using trypan blue. Alamar Blue was used to test drug sensitivity. Differential genes were screened by RNA sequencing. Reverse transcription‑quantitative PCR and western blotting were performed to assess the expressions of CSF2/p‑STAT3/MYC pathway related molecules, neuroendocrine (NE)/non‑neuroendocrine (non‑NE), transcription factors and drug‑related targets. The present study found that SCLC cell line NCI‑H69 exhibited adherent (H69A) and suspensive (H69S) phenotypes, which could switch back and forth. The two phenotypic cells had significant differences in cellular NE and non‑NE characteristics, drug sensitivity and expression of drug‑related targets. RNA sequencing showed that granulocyte‑macrophage colony‑stimulating factor [i.e., colony‑stimulating factor 2 (CSF2)] was the main differentially expressed gene between the two phenotypes and that H69A cells highly expressed CSF2. The inhibition of CSF2 promoted the transformation from H69A to H69S, increased drug sensitivity and NE marker expression and decreased the non‑NE marker expression in H69A. The STRING, Pathway Commons and Reactome databases showed a potential regulatory relationship between CSF2 and phosphorylated signal transducer and activator of transcription 3 (p‑STAT3)/MYC. p‑STAT3 and MYC expression was higher in H69A cells than in H69S cells and CSF2 silencing inhibited their expression. Taken together, these results indicated that CSF2 may regulate the phenotypic plasticity of SCLC through the phosphorylated STAT3/MYC pathway, thereby limiting the transformation between cell clones with different phenotypes and changing the sensitivity of specific cell clones to targeted drugs. Targeting CSF2 may be a potential therapeutic strategy to overcome drug resistance in SCLC treatment by influencing ITH.
Keywords: drug resistance; granulocyte‑macrophage colony‑stimulating factor; intratumoral heterogeneity; phenotypic plasticity; small cell lung cancer.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway.FEBS J. 2020 Dec;287(23):5218-5235. doi: 10.1111/febs.15311. Epub 2020 Apr 17. FEBS J. 2020. PMID: 32239647 Free PMC article.
-
The potential mechanism of WT1-associated protein-induced N-6-methyladenosine modification of colony-stimulating factor 2 in the progression of oral squamous cell carcinoma by JAK/STAT3 pathway regulation.Eur J Oral Sci. 2024 Aug;132(4):e13001. doi: 10.1111/eos.13001. Epub 2024 Jun 3. Eur J Oral Sci. 2024. PMID: 38831514
-
Dynamic phenotypic reprogramming and chemoresistance induced by lung fibroblasts in small cell lung cancer.Sci Rep. 2024 Feb 5;14(1):2884. doi: 10.1038/s41598-024-52687-z. Sci Rep. 2024. PMID: 38311608 Free PMC article.
-
Killing SCLC: insights into how to target a shapeshifting tumor.Genes Dev. 2022 Mar 1;36(5-6):241-258. doi: 10.1101/gad.349359.122. Genes Dev. 2022. PMID: 35318269 Free PMC article. Review.
-
Role of microRNAs in regulating cell proliferation, metastasis and chemoresistance and their applications as cancer biomarkers in small cell lung cancer.Biochim Biophys Acta Rev Cancer. 2021 Aug;1876(1):188552. doi: 10.1016/j.bbcan.2021.188552. Epub 2021 Apr 21. Biochim Biophys Acta Rev Cancer. 2021. PMID: 33892053 Review.
Cited by
-
Neutrophil estimation and prognosis analysis based on existing lung squamous cell carcinoma datasets: the development and validation of a prognosis prediction model.Transl Lung Cancer Res. 2024 Aug 31;13(8):2023-2037. doi: 10.21037/tlcr-24-411. Epub 2024 Aug 19. Transl Lung Cancer Res. 2024. PMID: 39263021 Free PMC article.
-
Norepinephrine induces anoikis resistance in high-grade serous ovarian cancer precursor cells.JCI Insight. 2024 Mar 8;9(5):e170961. doi: 10.1172/jci.insight.170961. JCI Insight. 2024. PMID: 38271085 Free PMC article.
-
First-line serplulimab plus chemotherapy in extensive-stage small-cell lung cancer: Updated results and biomarker analysis from the ASTRUM-005 randomized clinical trial.Cancer Commun (Lond). 2025 Aug;45(8):990-1009. doi: 10.1002/cac2.70032. Epub 2025 May 29. Cancer Commun (Lond). 2025. PMID: 40440184 Free PMC article. Clinical Trial.
-
T cell proliferation-related subtypes, prognosis model and characterization of tumor microenvironment in head and neck squamous cell carcinoma.Heliyon. 2024 Jul 10;10(14):e34221. doi: 10.1016/j.heliyon.2024.e34221. eCollection 2024 Jul 30. Heliyon. 2024. PMID: 39082023 Free PMC article.
-
[PKM1 Regulates the Expression of Autophagy and Neuroendocrine Markers in Small Cell Lung Cancer].Zhongguo Fei Ai Za Zhi. 2024 Sep 20;27(9):645-653. doi: 10.3779/j.issn.1009-3419.2024.102.33. Zhongguo Fei Ai Za Zhi. 2024. PMID: 39492579 Free PMC article. Chinese.
References
-
- Nicholson AG, Chansky K, Crowley J, Beyruti R, Kubota K, Turrisi A, Eberhardt WE, van Meerbeeck J, Rami-Porta R; Staging, Prognostic Factors Committee, et al. The international association for the study of lung cancer lung cancer staging project: Proposals for the revision of the clinical and pathologic staging of small cell lung cancer in the forthcoming eighth edition of the tnm classification for lung cancer. J Thorac Oncol. 2016;11:300–311. doi: 10.1016/j.jtho.2015.10.008. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
