

Human KCNQ5 de novo mutations underlie epilepsy and intellectual disability
- PMID: 35583973
- PMCID: PMC9236882
- DOI: 10.1152/jn.00509.2021
Human KCNQ5 de novo mutations underlie epilepsy and intellectual disability
Abstract
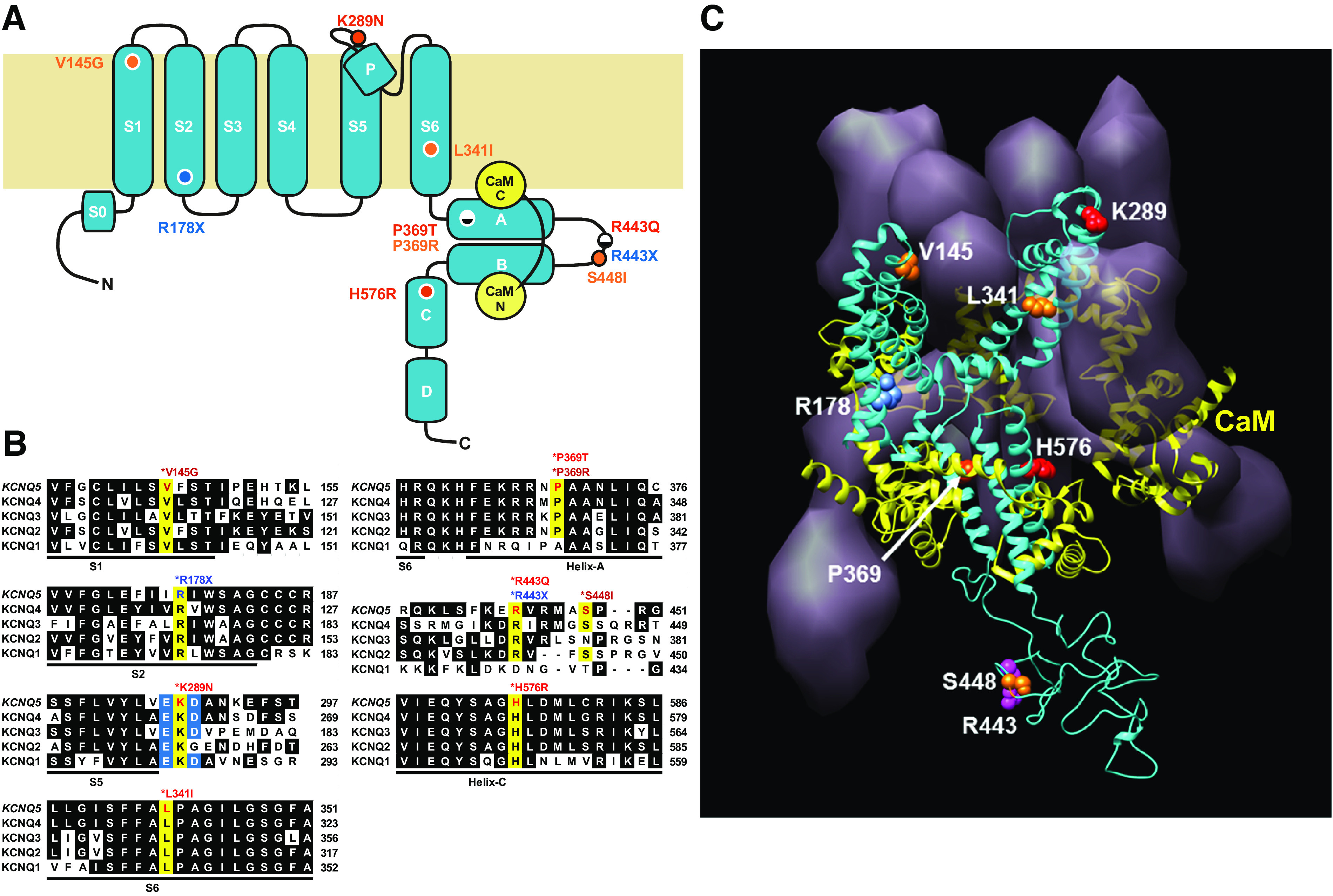
We identified six novel de novo human KCNQ5 variants in children with motor/language delay, intellectual disability (ID), and/or epilepsy by whole exome sequencing. These variants, comprising two nonsense and four missense alterations, were functionally characterized by electrophysiology in HEK293/CHO cells, together with four previously reported KCNQ5 missense variants (Lehman A, Thouta S, Mancini GM, Naidu S, van Slegtenhorst M, McWalter K, Person R, Mwenifumbo J, Salvarinova R; CAUSES Study; EPGEN Study; Guella I, McKenzie MB, Datta A, Connolly MB, Kalkhoran SM, Poburko D, Friedman JM, Farrer MJ, Demos M, Desai S, Claydon T. Am J Hum Genet 101: 65-74, 2017). Surprisingly, all eight missense variants resulted in gain of function (GOF) due to hyperpolarized voltage dependence of activation or slowed deactivation kinetics, whereas the two nonsense variants were confirmed to be loss of function (LOF). One severe GOF allele (P369T) was tested and found to extend a dominant GOF effect to heteromeric KCNQ5/3 channels. Clinical presentations were associated with altered KCNQ5 channel gating: milder presentations with LOF or smaller GOF shifts in voltage dependence [change in voltage at half-maximal conduction (ΔV50) = ∼-15 mV] and severe presentations with larger GOF shifts in voltage dependence (ΔV50 = ∼-30 mV). To examine LOF pathogenicity, two Kcnq5 LOF mouse lines were created with CRISPR/Cas9. Both lines exhibited handling- and thermal-induced seizures and abnormal cortical EEGs consistent with epileptiform activity. Our study thus provides evidence for in vivo KCNQ5 LOF pathogenicity and strengthens the contribution of both LOF and GOF mutations to global pediatric neurological impairment, including ID/epilepsy.NEW & NOTEWORTHY Six novel de novo human KCNQ5 variants were identified from children with neurodevelopmental delay, intellectual disability, and/or epilepsy. Expression of these variants along with four previously reported KCNQ5 variants from a similar cohort revealed GOF potassium channels, negatively shifted in V50 of activation and/or delayed deactivation kinetics. GOF is extended to KCNQ5/3 heteromeric channels, making these the predominant channels affected in heterozygous de novo patients. Kcnq5 LOF mice exhibited seizures, consistent with in vivo pathogenicity.
Keywords: KCNQ5; M current; channelopathy; epilepsy; intellectual disability.
Conflict of interest statement
D.N.S. and K.L.H were employed by Ambry Genetics during this study. J.-M. R.is an editor of the
Jan-Marino Ramirez is an editor of
Figures










References
-
- Alexander SP, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA; CGTP Collaborators. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: voltage-gated ion channels. Br J Pharmacol 174, Suppl 1: S160–S194, 2017. doi: 10.1111/bph.13884. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous