

The Bradycardic Agent Ivabradine Acts as an Atypical Inhibitor of Voltage-Gated Sodium Channels
- PMID: 35586063
- PMCID: PMC9108390
- DOI: 10.3389/fphar.2022.809802
The Bradycardic Agent Ivabradine Acts as an Atypical Inhibitor of Voltage-Gated Sodium Channels
Abstract
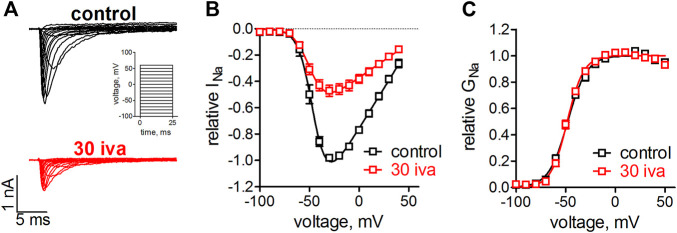
Background and purpose: Ivabradine is clinically administered to lower the heart rate, proposedly by inhibiting hyperpolarization-activated cyclic nucleotide-gated cation channels in the sinoatrial node. Recent evidence suggests that voltage-gated sodium channels (VGSC) are inhibited within the same concentration range. VGSCs are expressed within the sinoatrial node and throughout the conduction system of the heart. A block of these channels thus likely contributes to the established and newly raised clinical indications of ivabradine. We, therefore, investigated the pharmacological action of ivabradine on VGSCs in sufficient detail in order to gain a better understanding of the pro- and anti-arrhythmic effects associated with the administration of this drug. Experimental Approach: Ivabradine was tested on VGSCs in native cardiomyocytes isolated from mouse ventricles and the His-Purkinje system and on human Nav1.5 in a heterologous expression system. We investigated the mechanism of channel inhibition by determining its voltage-, frequency-, state-, and temperature-dependence, complemented by a molecular drug docking to the recent Nav1.5 cryoEM structure. Automated patch-clamp experiments were used to investigate ivabradine-mediated changes in Nav1.5 inactivation parameters and inhibition of different VGSC isoforms. Key results: Ivabradine inhibited VGSCs in a voltage- and frequency-dependent manner, but did not alter voltage-dependence of activation and fast inactivation, nor recovery from fast inactivation. Cardiac (Nav1.5), neuronal (Nav1.2), and skeletal muscle (Nav1.4) VGSC isoforms were inhibited by ivabradine within the same concentration range, as were sodium currents in native cardiomyocytes isolated from the ventricles and the His-Purkinje system. Molecular drug docking suggested an interaction of ivabradine with the classical local anesthetic binding site. Conclusion and Implications: Ivabradine acts as an atypical inhibitor of VGSCs. Inhibition of VGSCs likely contributes to the heart rate lowering effect of ivabradine, in particular at higher stimulation frequencies and depolarized membrane potentials, and to the observed slowing of intra-cardiac conduction. Inhibition of VGSCs in native cardiomyocytes and across channel isoforms may provide a potential basis for the anti-arrhythmic potential as observed upon administration of ivabradine.
Keywords: S16257; atypical inhibitor; conduction cell; ivabradine; ventricular cardiomyocyte; voltage-gated sodium channel.
Copyright © 2022 Hackl, Lukacs, Ebner, Pesti, Haechl, Földi, Lilliu, Schicker, Kubista, Stary-Weinzinger, Hilber, Mike, Todt and Koenig.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures








References
-
- Abraham S., Beatch G. N., MacLeod B. A., Walker M. J. (1989). Antiarrhythmic Properties of Tetrodotoxin against Occlusion-Induced Arrhythmias in the Rat: A Novel Approach to the Study of the Antiarrhythmic Effects of Ventricular Sodium Channel Blockade. J. Pharmacol. Exp. Ther. 251, 1166–1173. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources