

Mixing genome annotation methods in a comparative analysis inflates the apparent number of lineage-specific genes
- PMID: 35588743
- PMCID: PMC9346927
- DOI: 10.1016/j.cub.2022.04.085
Mixing genome annotation methods in a comparative analysis inflates the apparent number of lineage-specific genes
Abstract
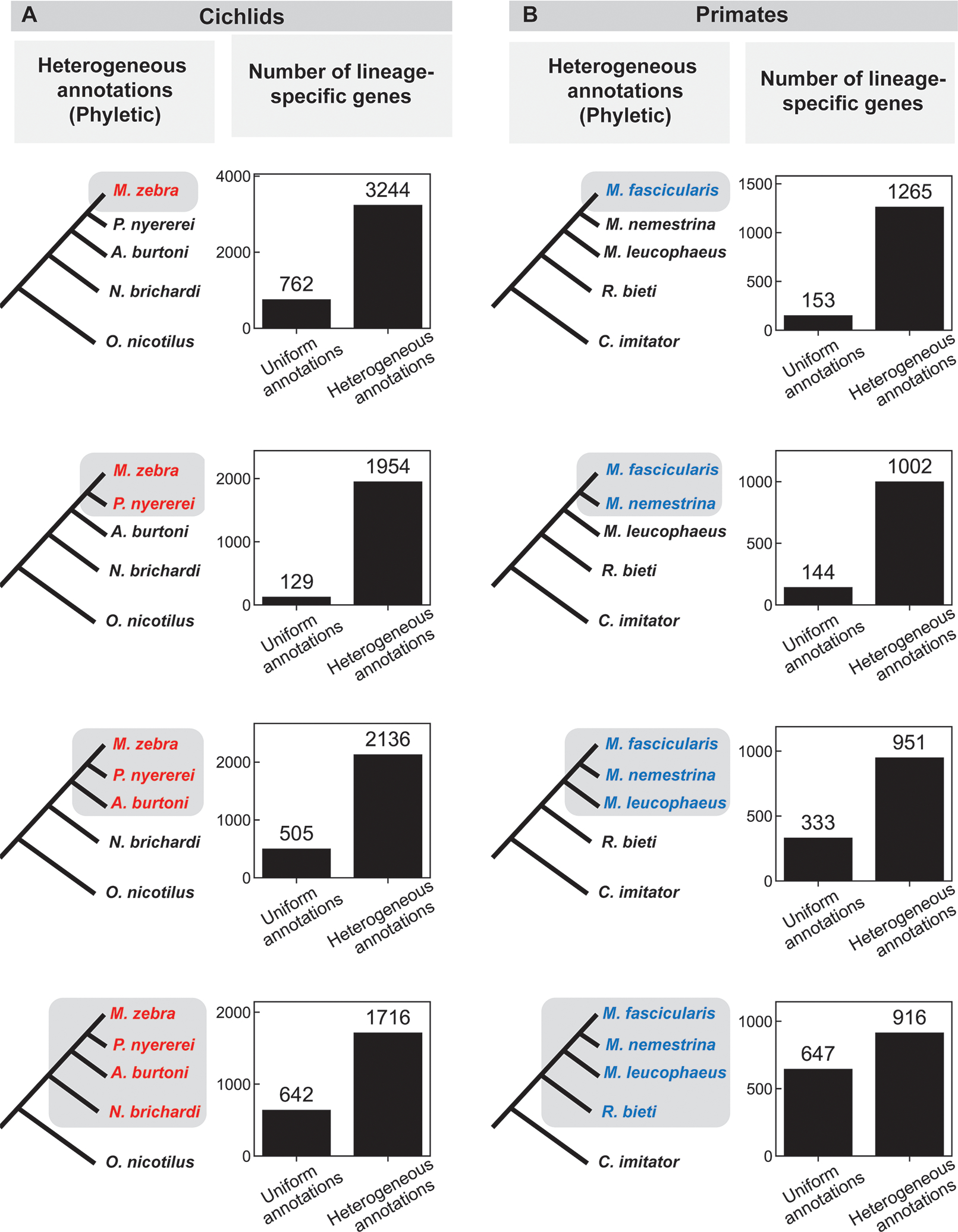
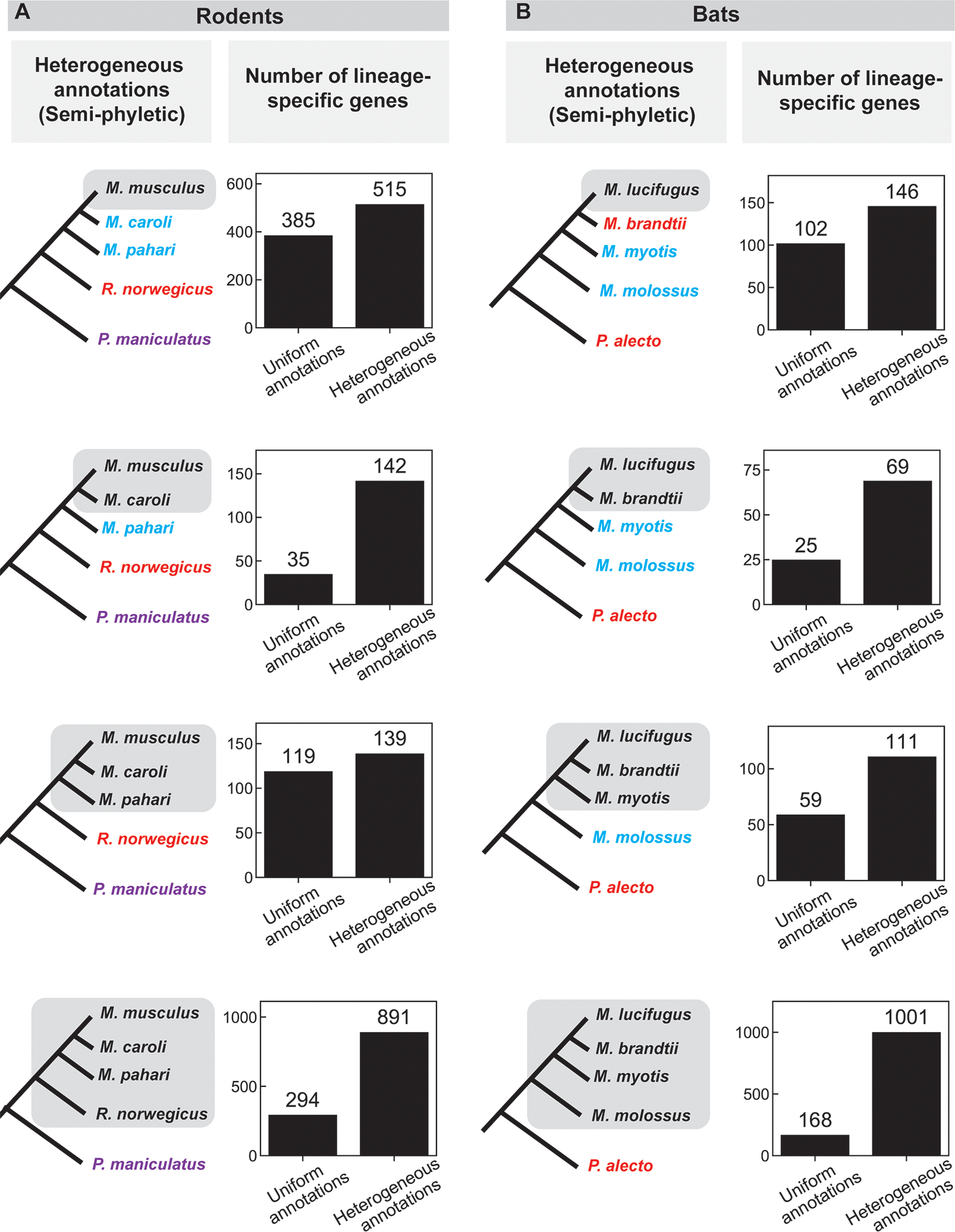
Comparisons of genomes of different species are used to identify lineage-specific genes, those genes that appear unique to one species or clade. Lineage-specific genes are often thought to represent genetic novelty that underlies unique adaptations. Identification of these genes depends not only on genome sequences, but also on inferred gene annotations. Comparative analyses typically use available genomes that have been annotated using different methods, increasing the risk that orthologous DNA sequences may be erroneously annotated as a gene in one species but not another, appearing lineage specific as a result. To evaluate the impact of such "annotation heterogeneity," we identified four clades of species with sequenced genomes with more than one publicly available gene annotation, allowing us to compare the number of lineage-specific genes inferred when differing annotation methods are used to those resulting when annotation method is uniform across the clade. In these case studies, annotation heterogeneity increases the apparent number of lineage-specific genes by up to 15-fold, suggesting that annotation heterogeneity is a substantial source of potential artifact.
Keywords: genome annotation; lineage-specific genes; novel genes; orphan genes; taxonomically restricted genes.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Khalturin K, Hemmrich G, Fraune S, Augustin R, and Bosch TC (2009). More than just orphans: are taxonomically-restricted genes important in evolution? Trends in Genetics 25, 404–413. - PubMed
-
- Tautz D, and Domazet-Lošo T (2011). The evolutionary origin of orphan genes. Nature Reviews Genetics 12, 692. - PubMed
-
- Wilson G, Bertrand N, Patel Y, Hughes J, Feil E, and Field D (2005). Orphans as taxonomically restricted and ecologically important genes. Microbiology 151, 2499–2501. - PubMed
-
- Tautz D (2014). The discovery of de novo gene evolution. Perspectives in biology and medicine 57, 149–161. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
