

PU.1 and MYC transcriptional network defines synergistic drug responses to KIT and LSD1 inhibition in acute myeloid leukemia
- PMID: 35590033
- PMCID: PMC9256806
- DOI: 10.1038/s41375-022-01594-1
PU.1 and MYC transcriptional network defines synergistic drug responses to KIT and LSD1 inhibition in acute myeloid leukemia
Abstract
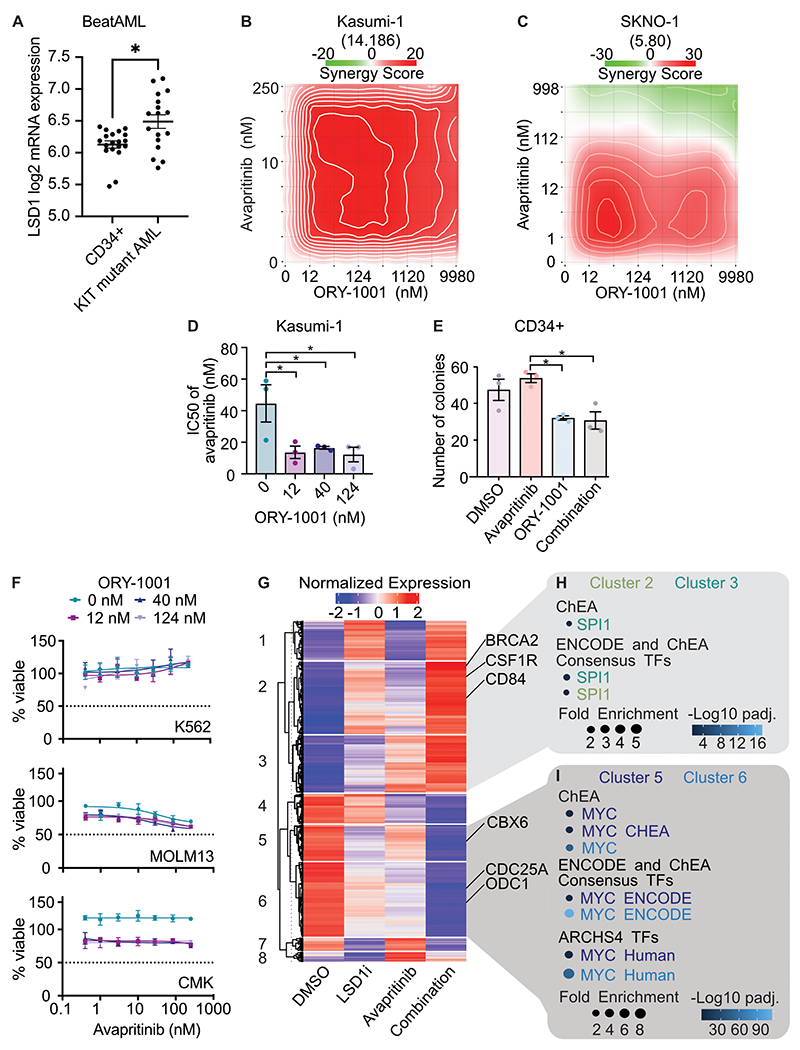
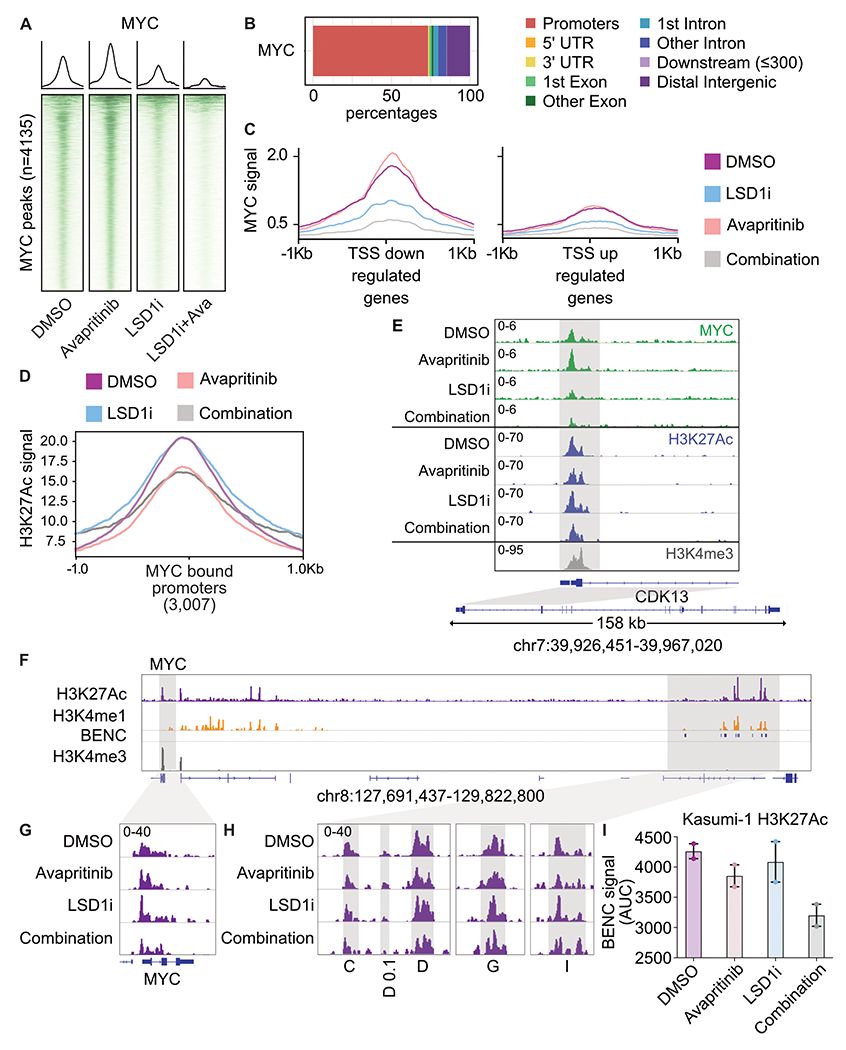
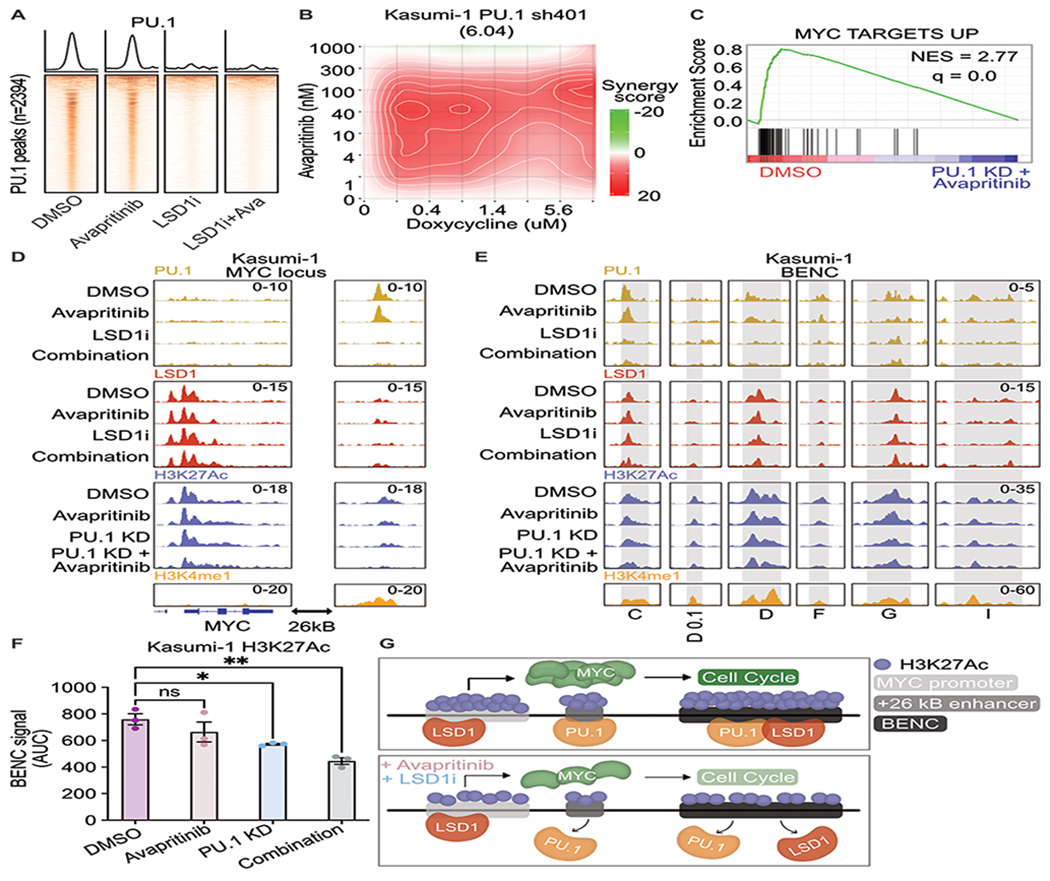
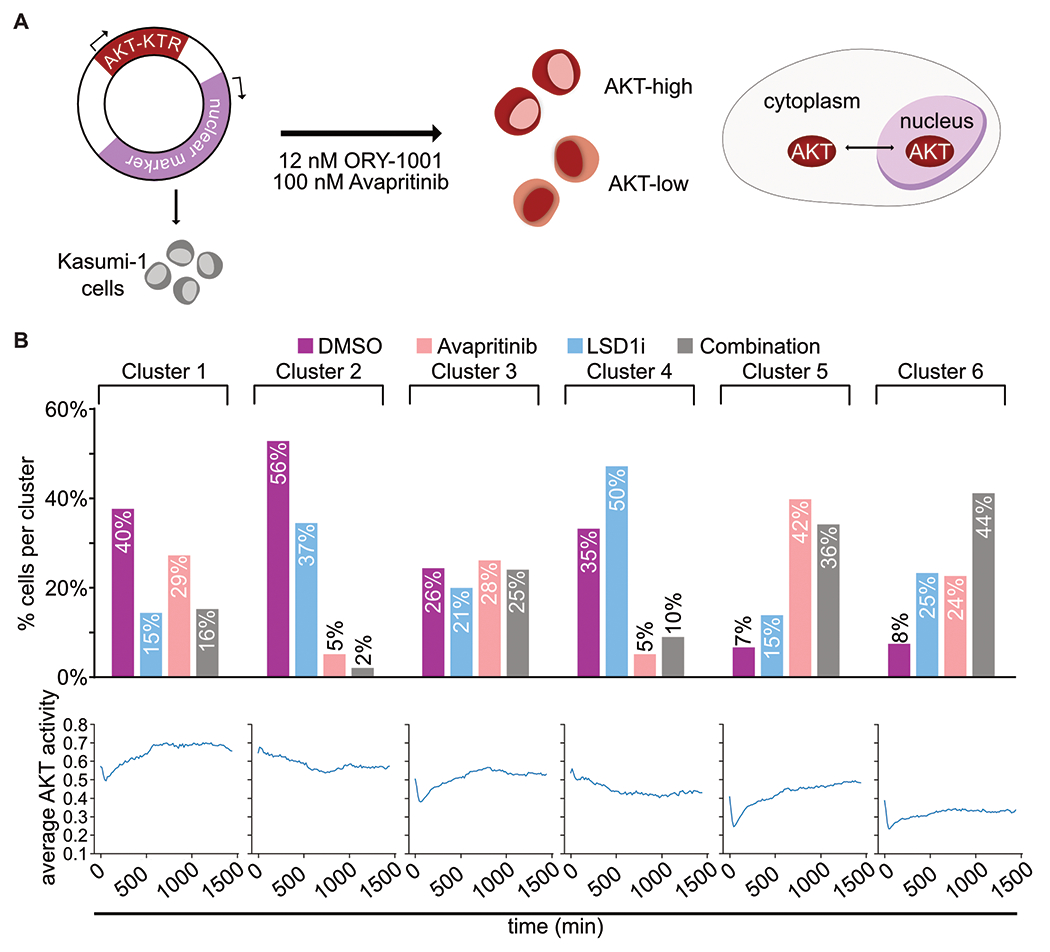
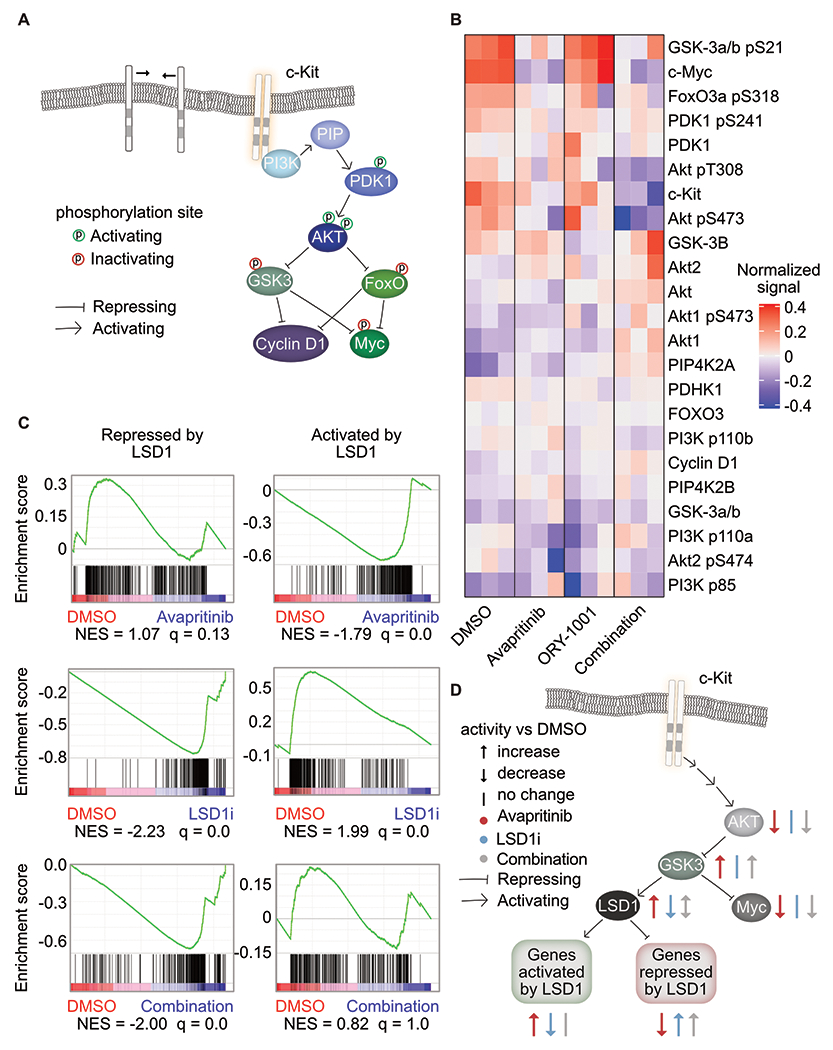
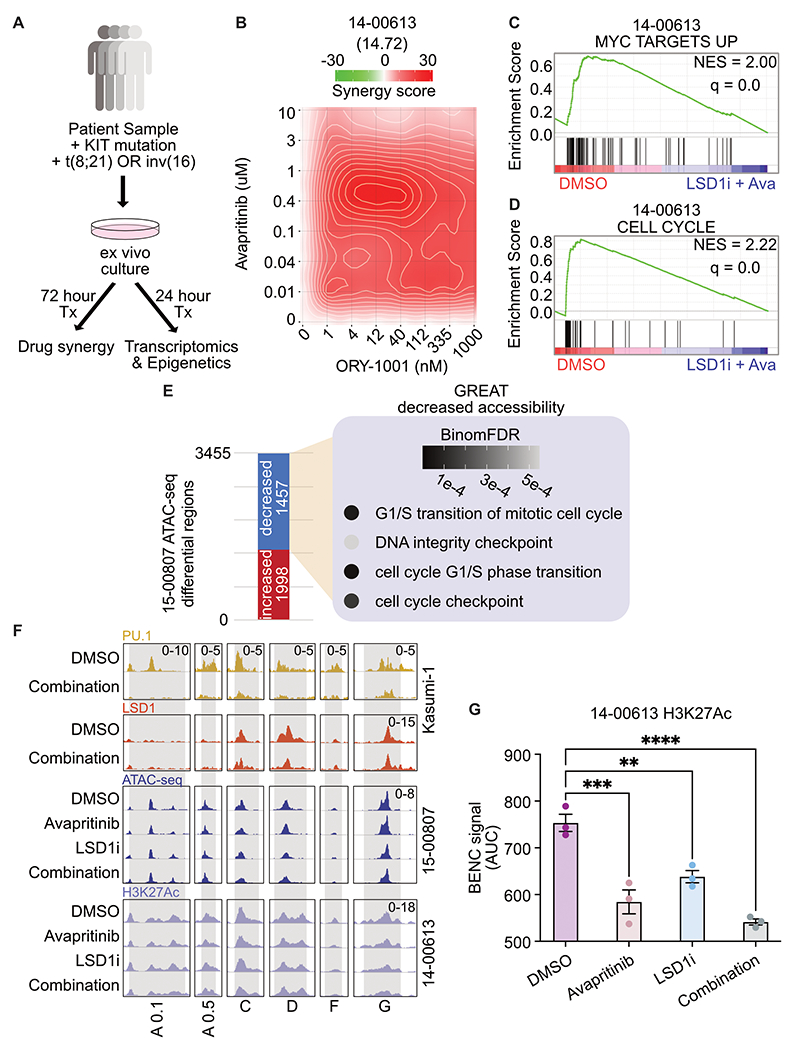
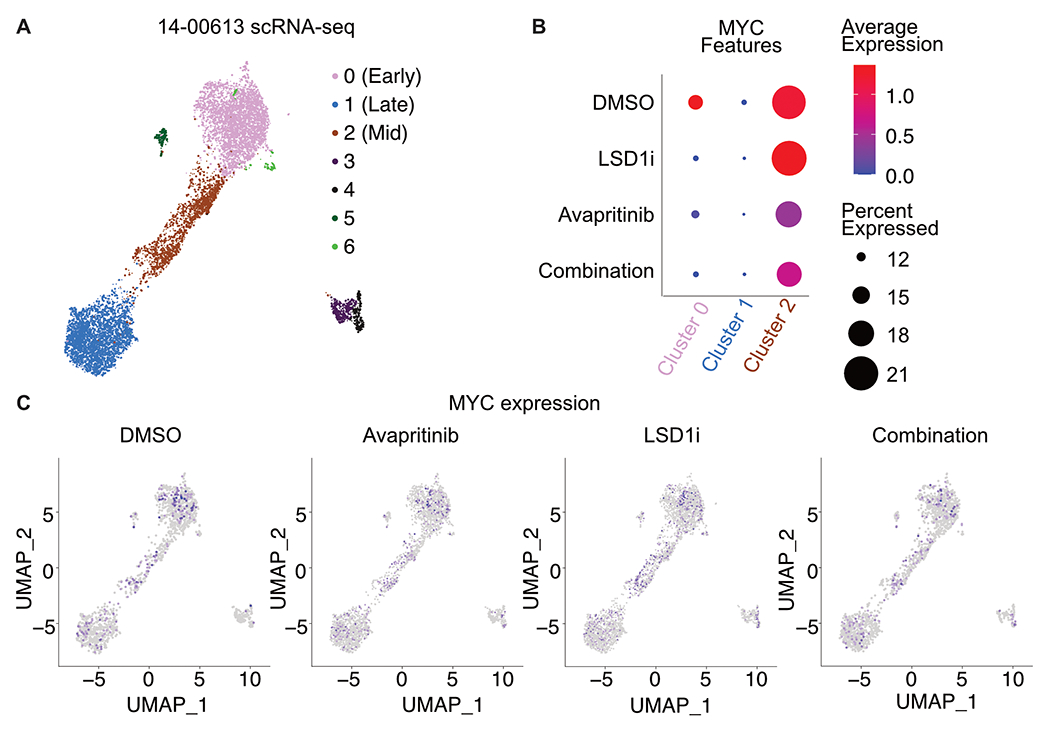
Responses to kinase-inhibitor therapy in AML are frequently short-lived due to the rapid development of resistance, limiting the clinical efficacy. Combination therapy may improve initial therapeutic responses by targeting pathways used by leukemia cells to escape monotherapy. Here we report that combined inhibition of KIT and lysine-specific demethylase 1 (LSD1) produces synergistic cell death in KIT-mutant AML cell lines and primary patient samples. This drug combination evicts both MYC and PU.1 from chromatin driving cell cycle exit. Using a live cell biosensor for AKT activity, we identify early adaptive changes in kinase signaling following KIT inhibition that are reversed with the addition of LSD1 inhibitor via modulation of the GSK3a/b axis. Multi-omic analyses, including scRNA-seq, ATAC-seq and CUT&Tag, confirm these mechanisms in primary KIT-mutant AML. Collectively, this work provides rational for a clinical trial to assess the efficacy of KIT and LSD1 inhibition in patients with KIT-mutant AML.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
References
-
- Salamero O, Montesinos P, Willekens C, Pérez-Simón JA, Pigneux A, Récher C et al. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J Clin Oncol 2020; 38: 4260–4273. - PMC - PubMed
-
- Odenike O, Wolff JE, Borthakur G, Aldoss IT, Rizzieri D, Prebet T et al. Results from the first-in-human study of mivebresib (ABBV-075), a pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol 2019; 37: 7030–7030.
-
- Dombret H, Preudhomme C, Berthon C, Raffoux E, Thomas X, Vey N et al. A Phase 1 Study of the BET-Bromodomain Inhibitor OTX015 in Patients with Advanced Acute Leukemia. Blood 2014; 124: 117–117.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
