

Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk
- PMID: 35590109
- PMCID: PMC9203933
- DOI: 10.1038/s41588-022-01069-0
Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk
Erratum in
-
Author Correction: Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk.Nat Genet. 2022 Aug;54(8):1259. doi: 10.1038/s41588-022-01142-8. Nat Genet. 2022. PMID: 35768727 No abstract available.
Abstract
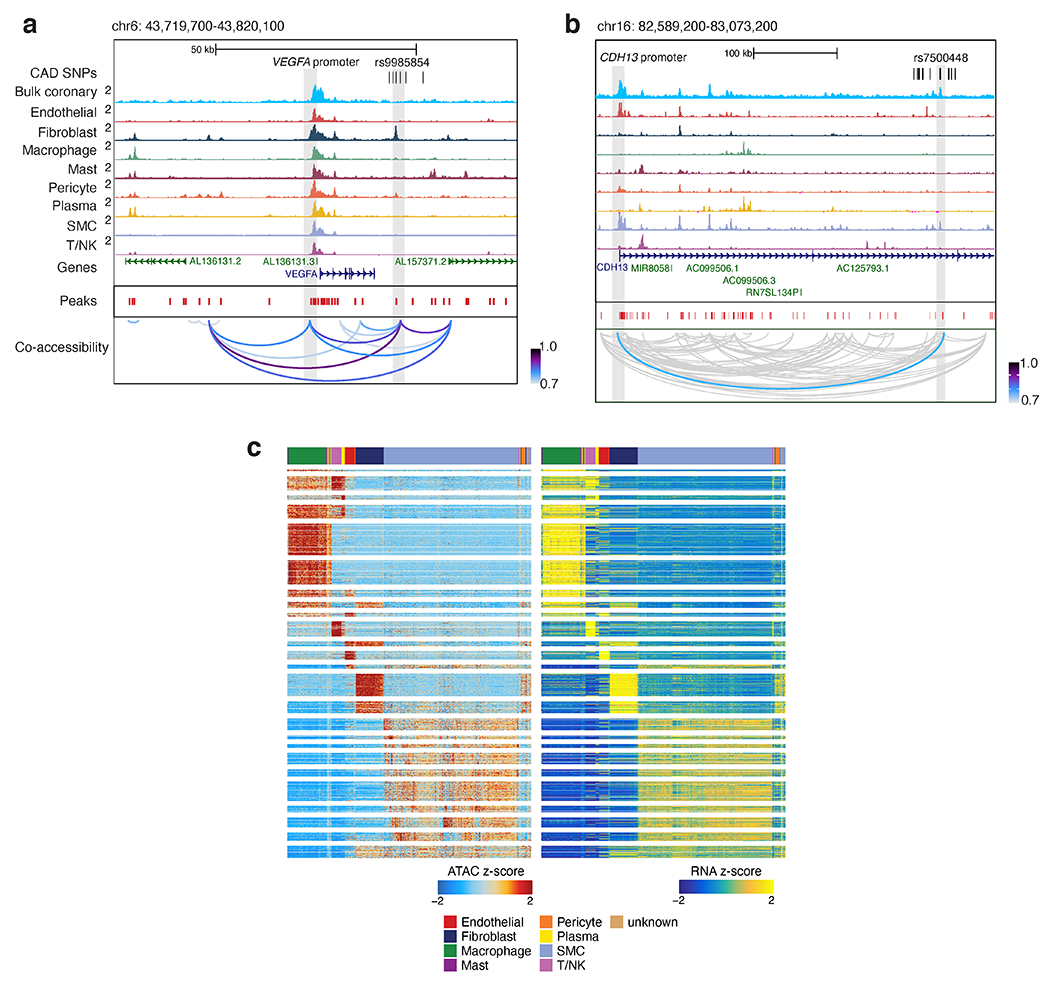
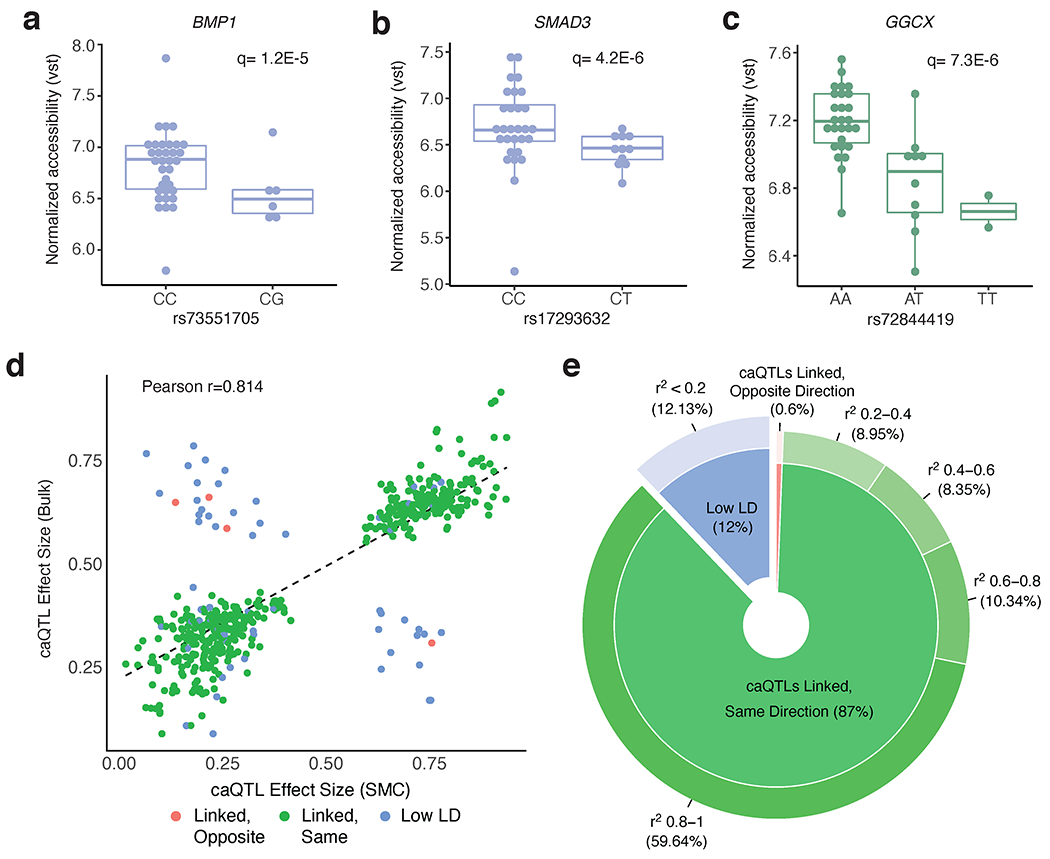
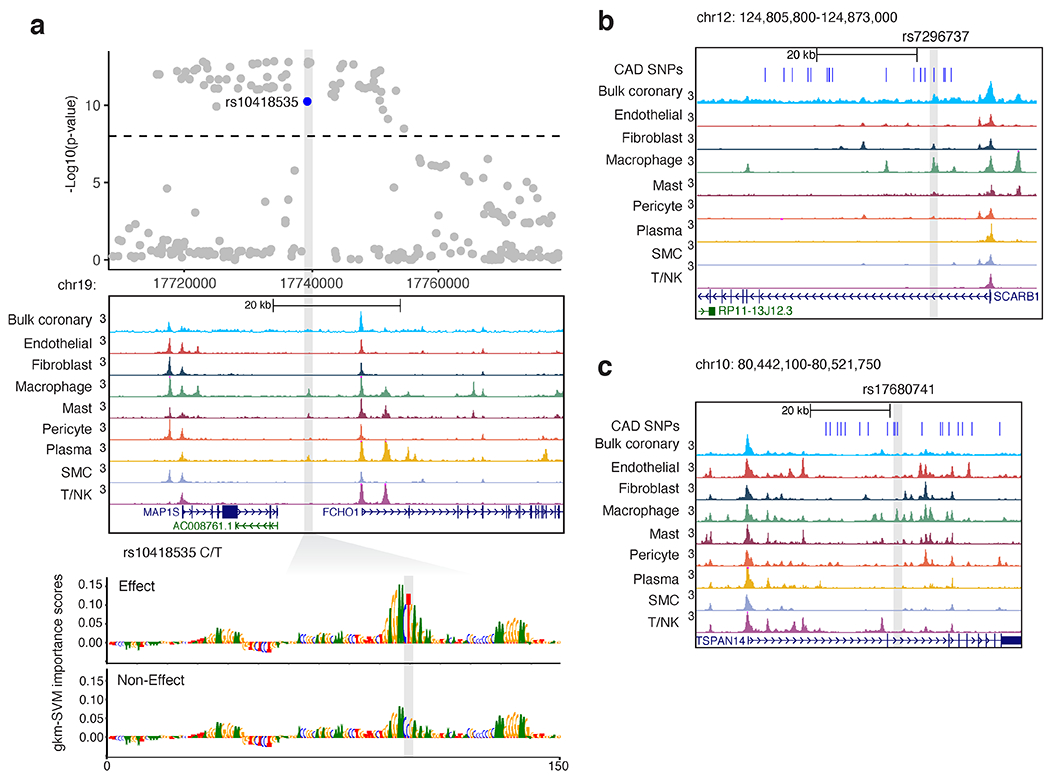
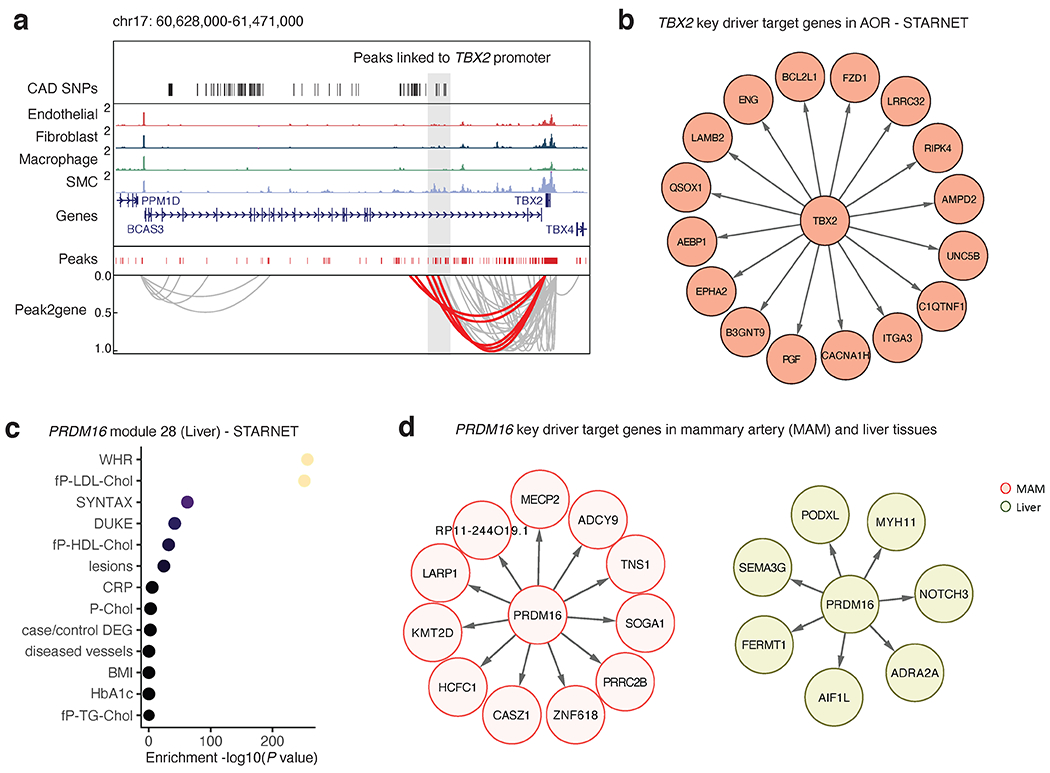
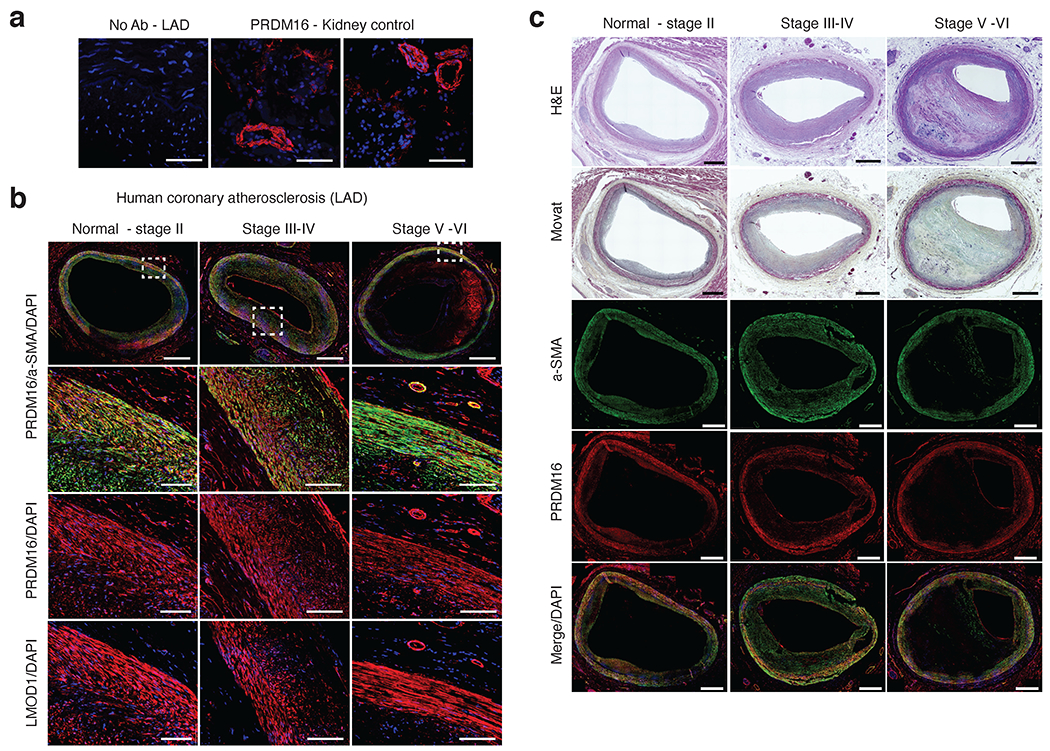
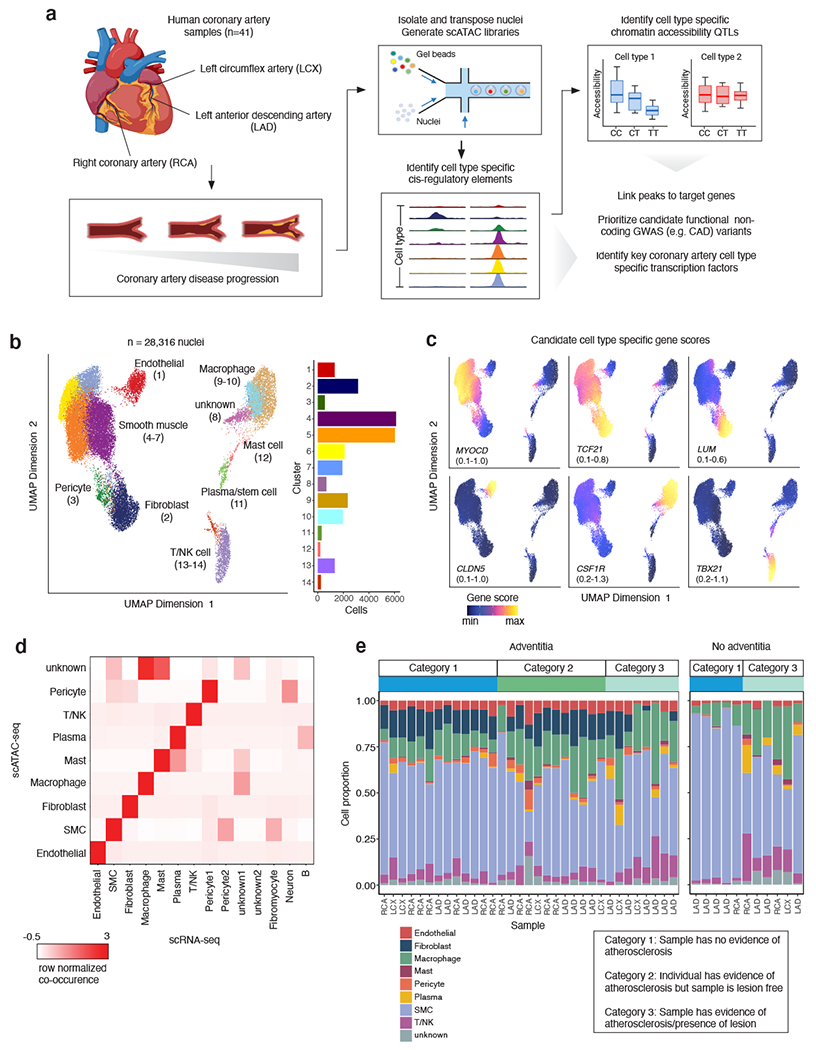
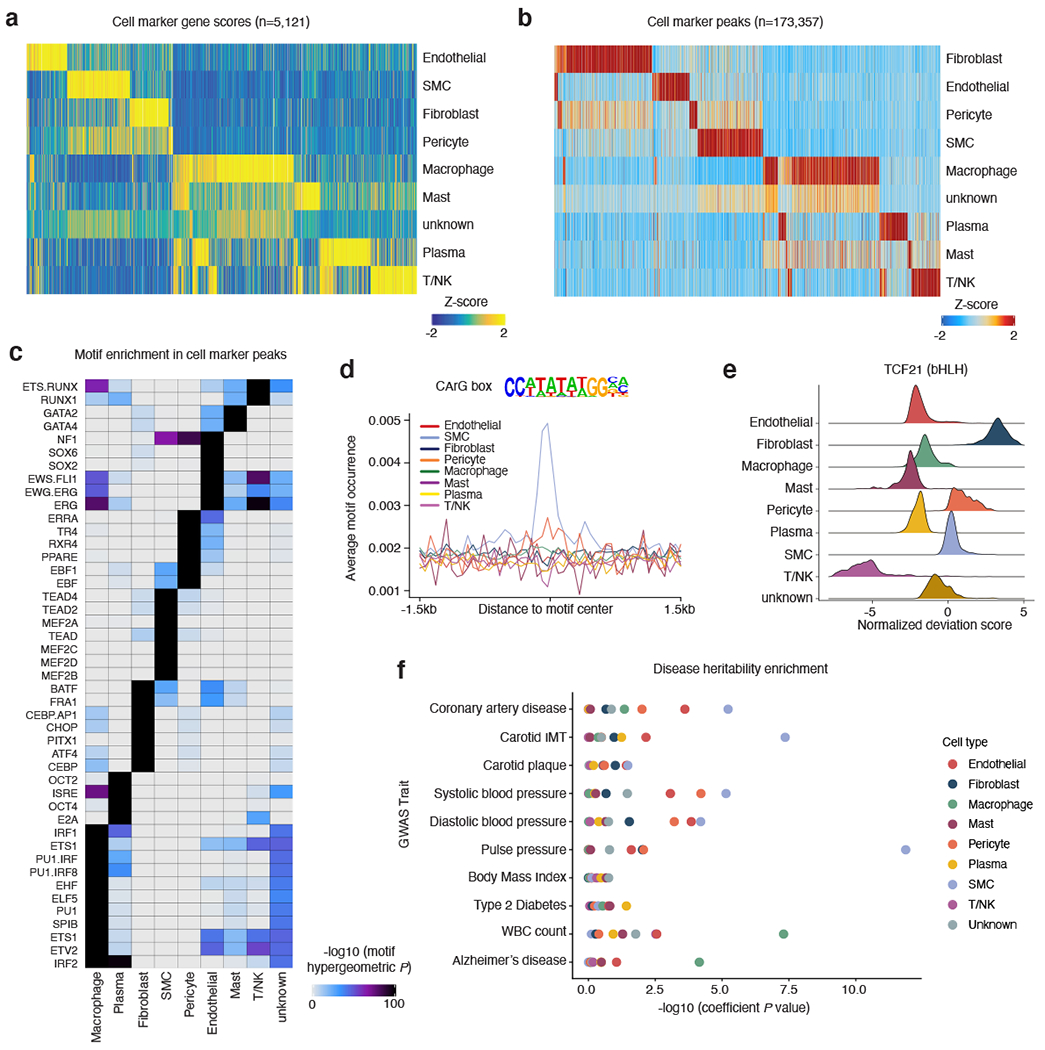
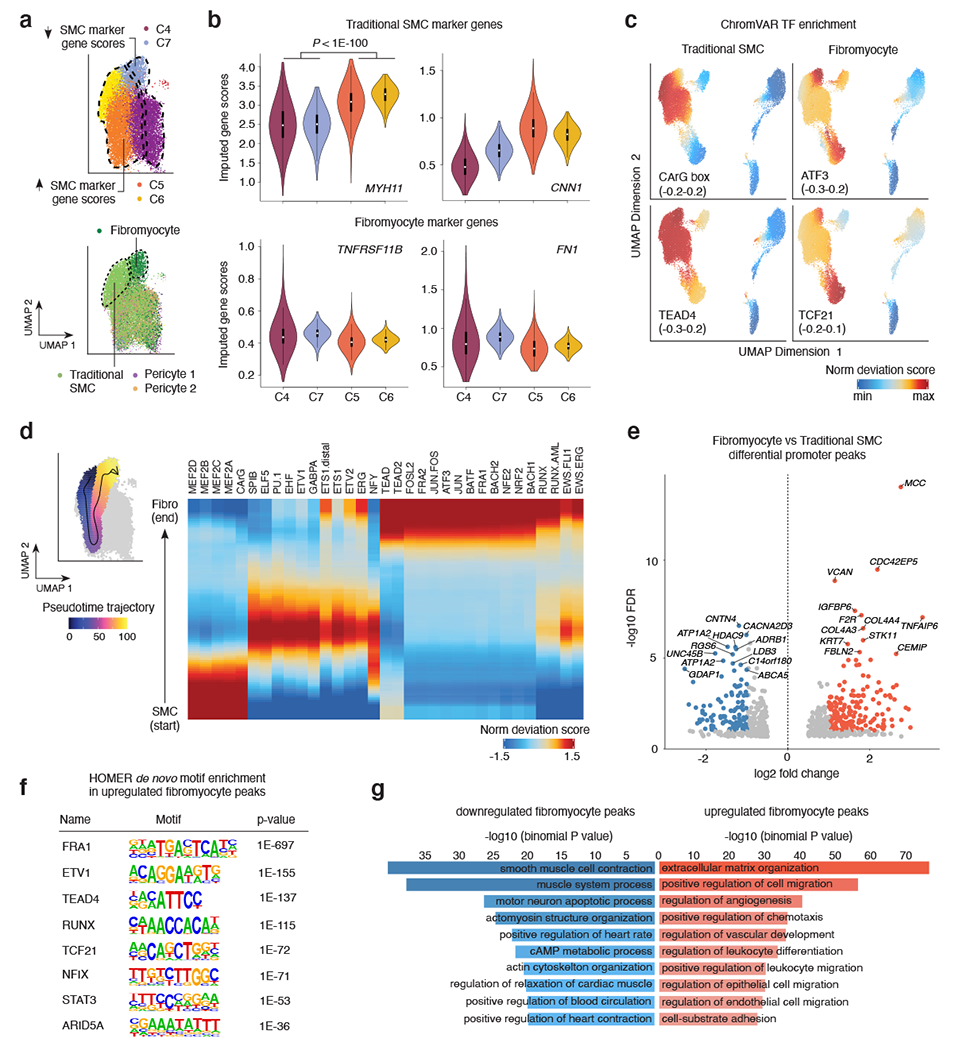
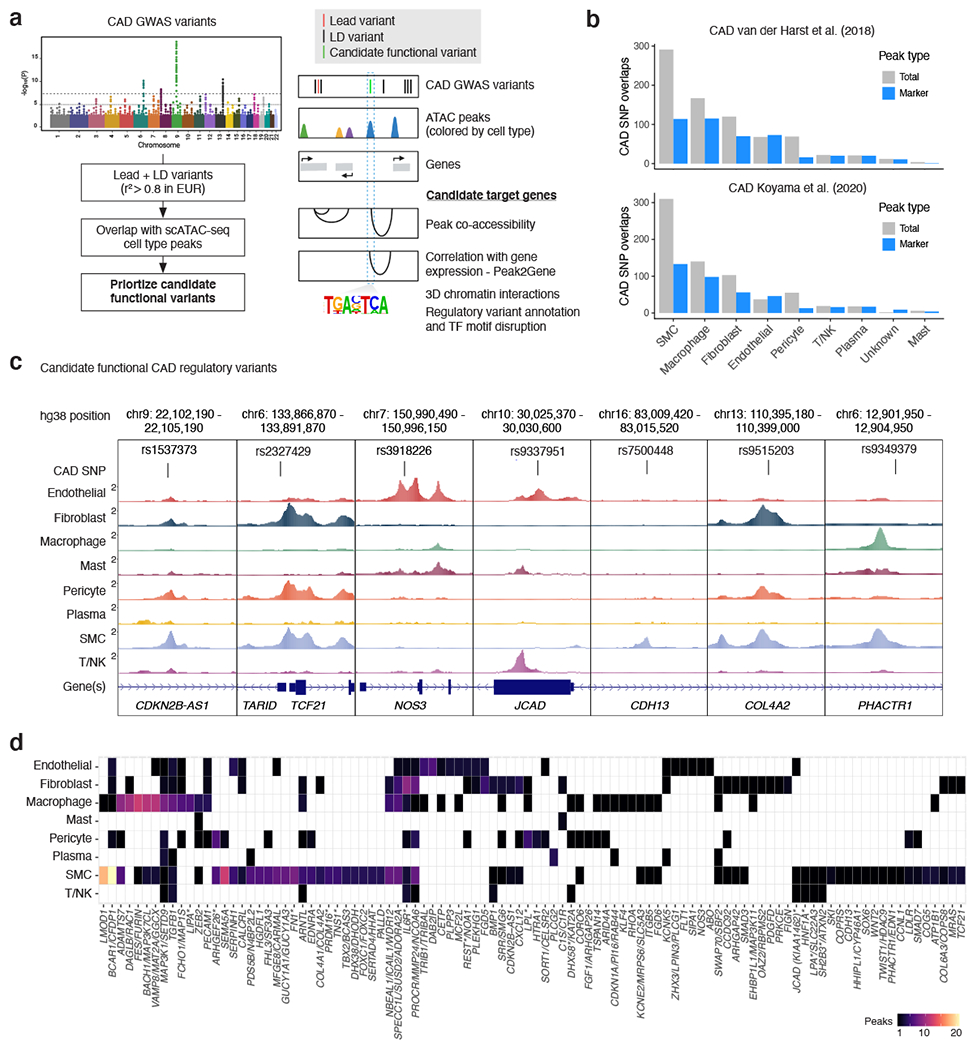
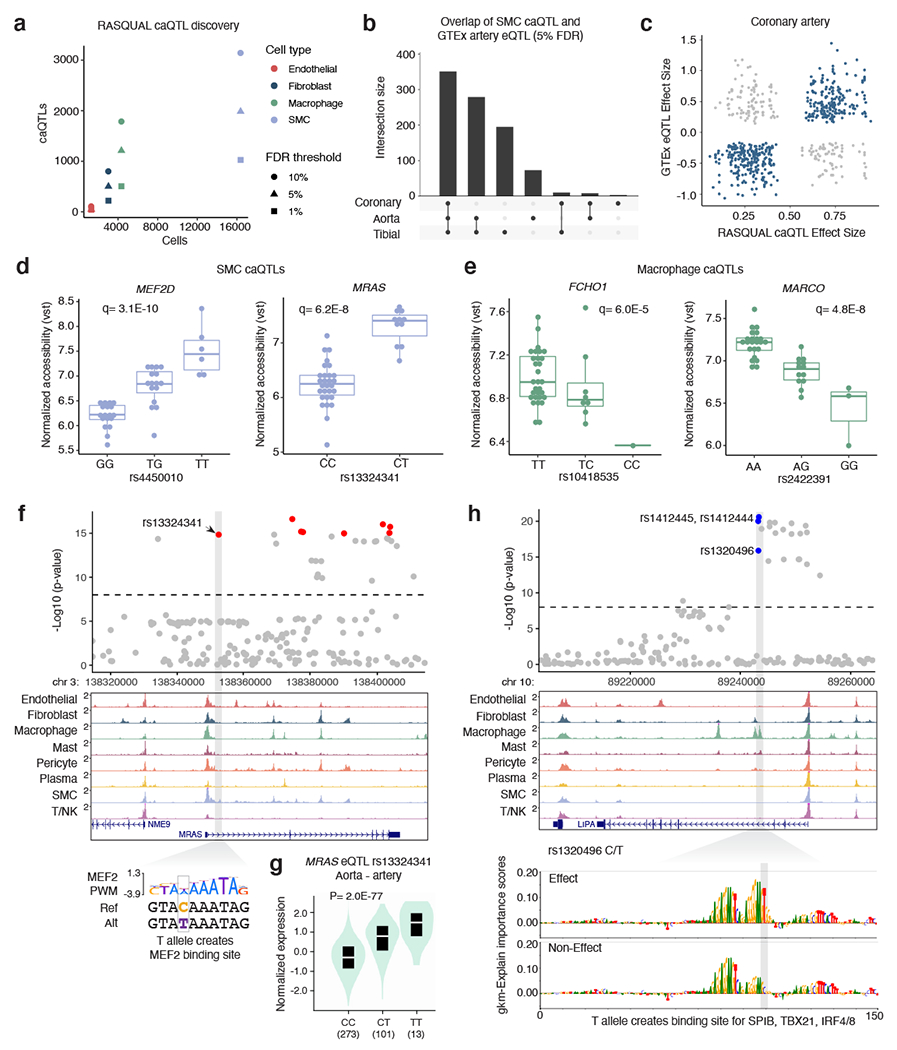
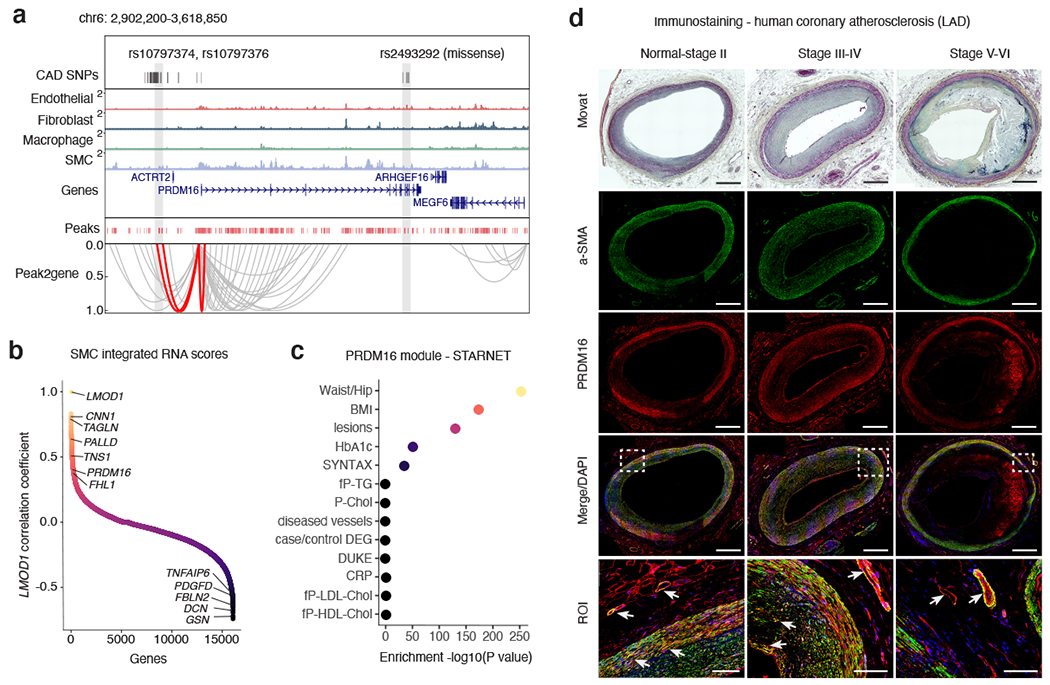
Coronary artery disease (CAD) is a complex inflammatory disease involving genetic influences across cell types. Genome-wide association studies have identified over 200 loci associated with CAD, where the majority of risk variants reside in noncoding DNA sequences impacting cis-regulatory elements. Here, we applied single-nucleus assay for transposase-accessible chromatin with sequencing to profile 28,316 nuclei across coronary artery segments from 41 patients with varying stages of CAD, which revealed 14 distinct cellular clusters. We mapped ~320,000 accessible sites across all cells, identified cell-type-specific elements and transcription factors, and prioritized functional CAD risk variants. We identified elements in smooth muscle cell transition states (for example, fibromyocytes) and functional variants predicted to alter smooth muscle cell- and macrophage-specific regulation of MRAS (3q22) and LIPA (10q23), respectively. We further nominated key driver transcription factors such as PRDM16 and TBX2. Together, this single-nucleus atlas provides a critical step towards interpreting regulatory mechanisms across the continuum of CAD risk.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
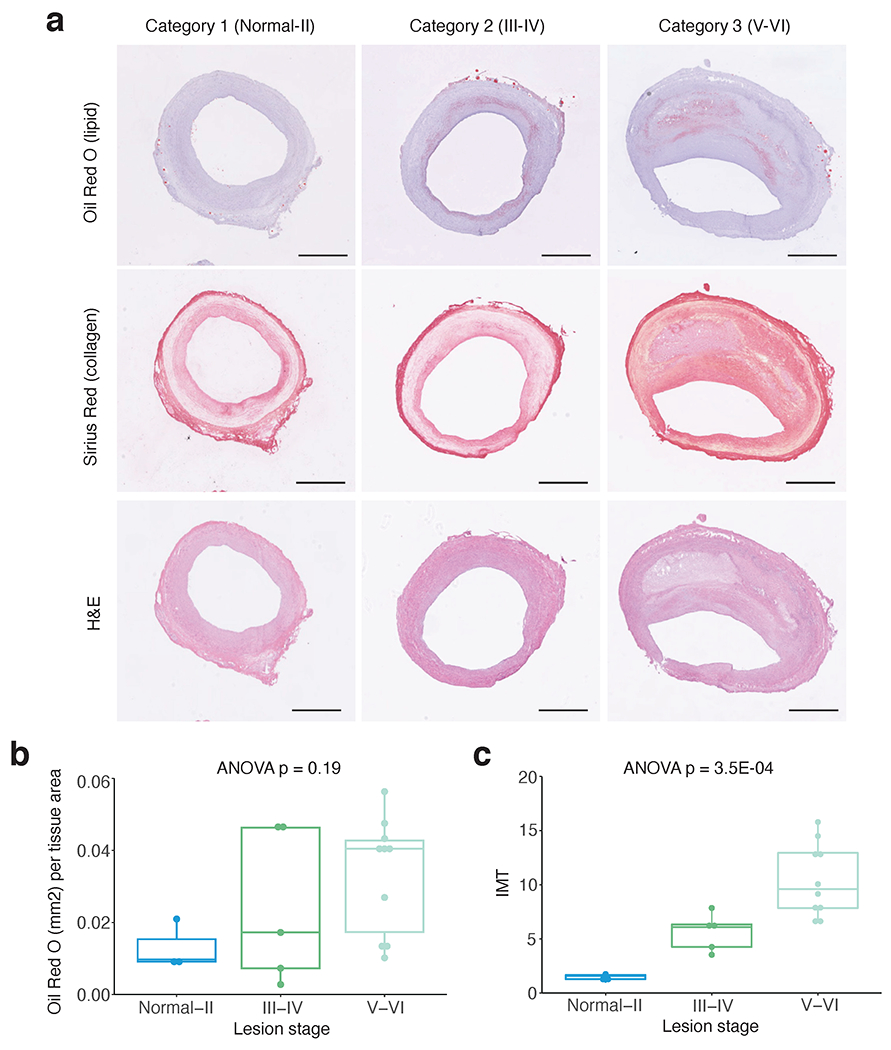
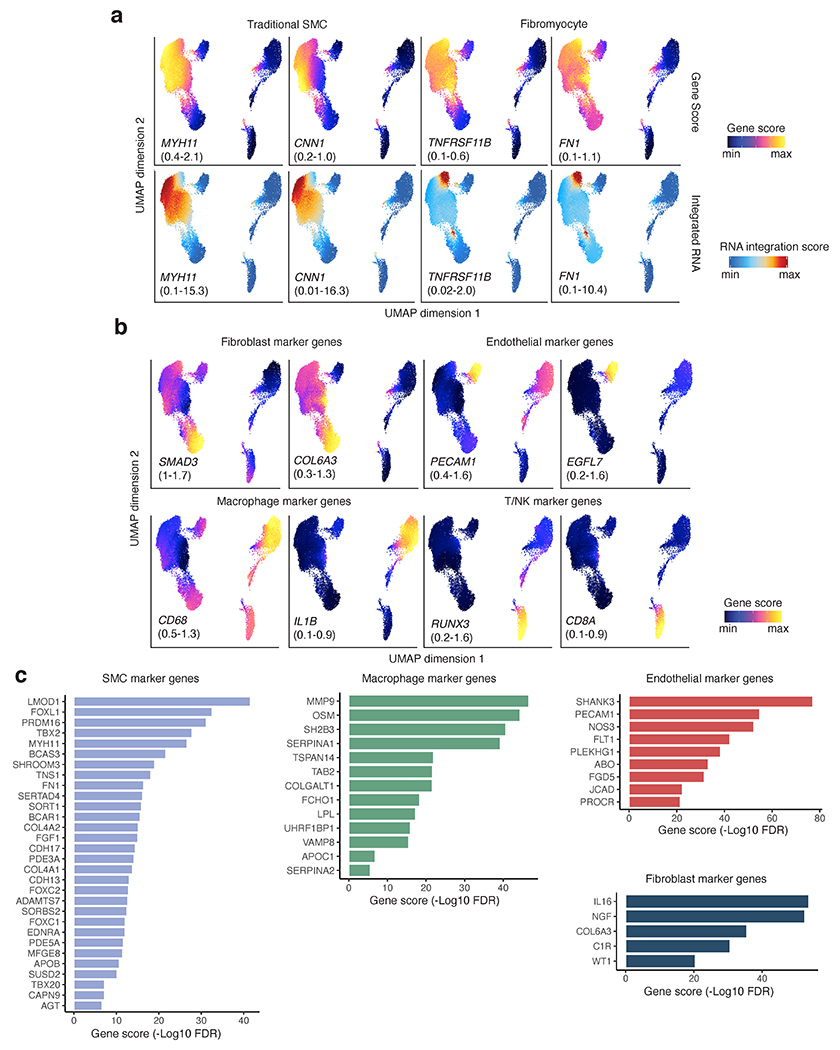
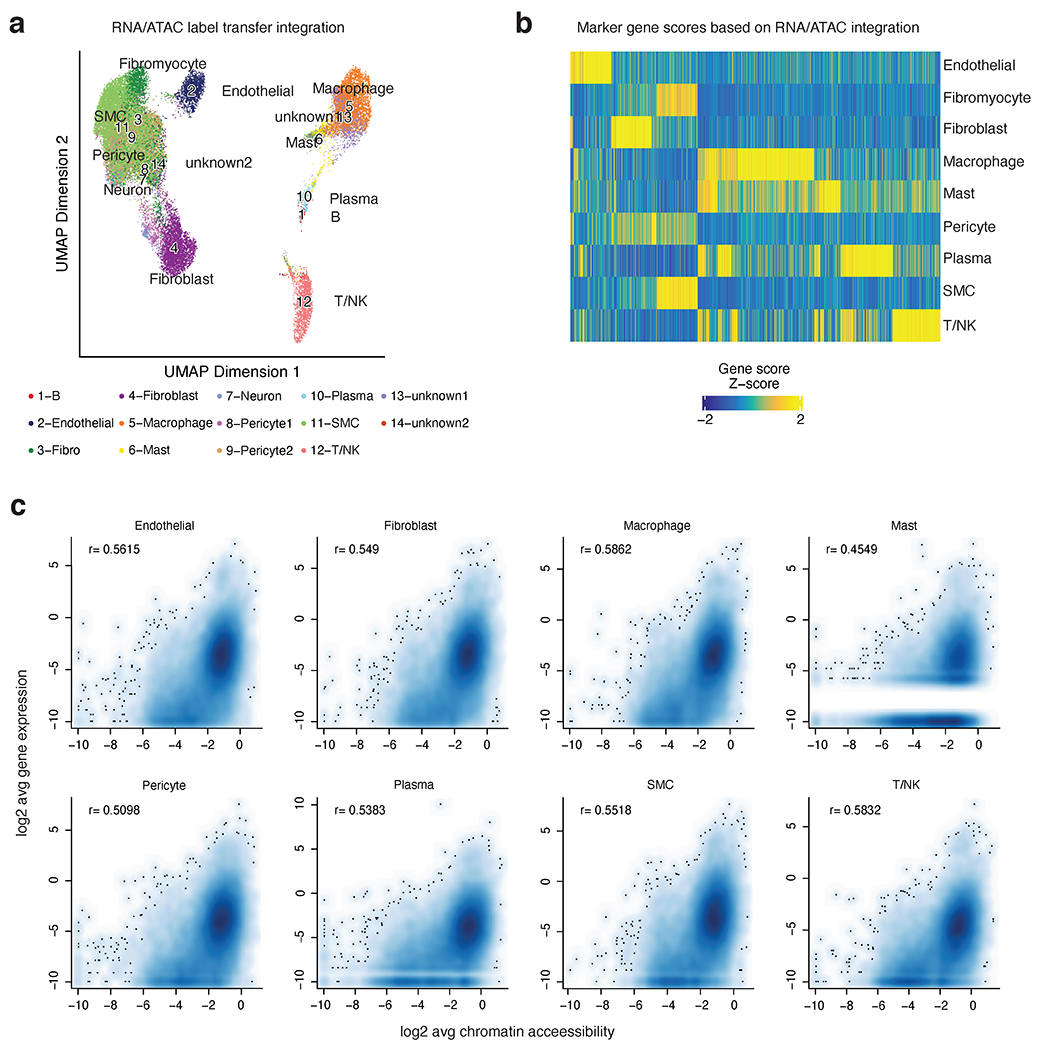
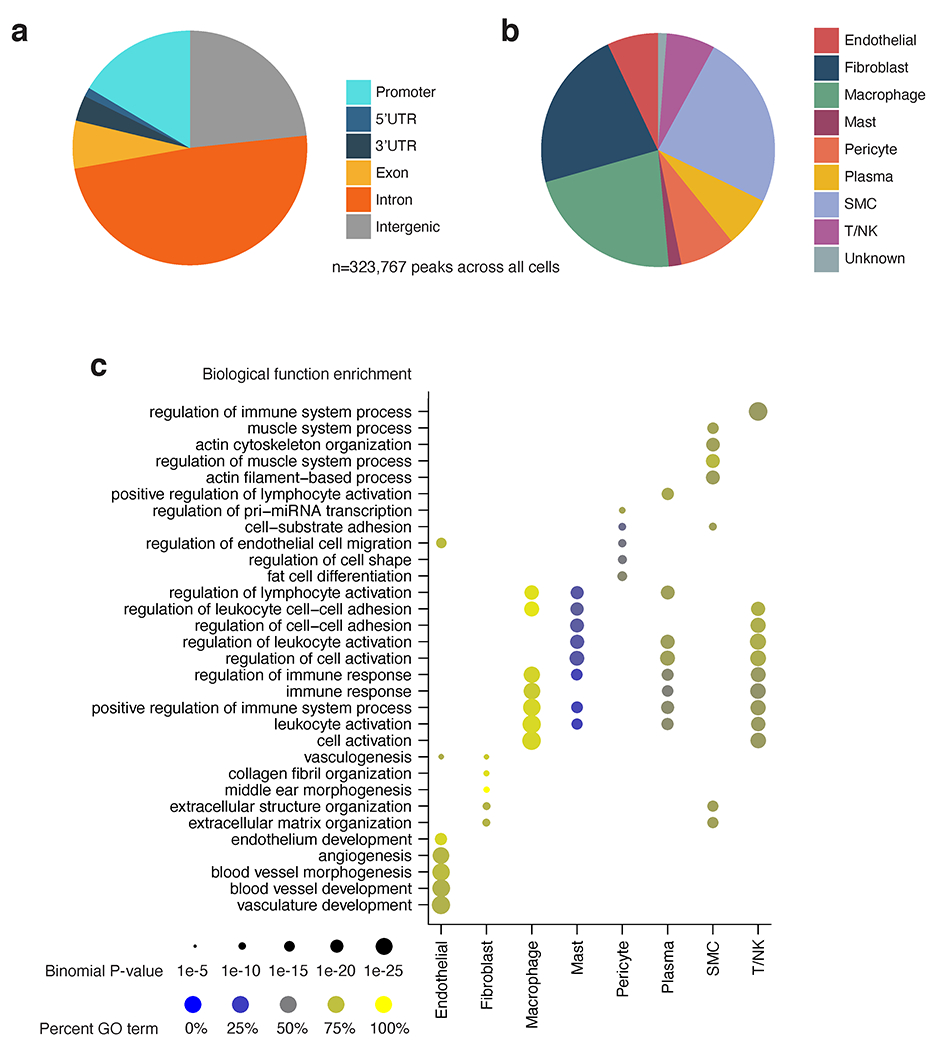
References
-
- Libby P Inflammation in atherosclerosis. Nature 420, 868–874 (2002). - PubMed
-
- Souilhol C, Harmsen MC, Evans PC & Krenning G Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res 114, 565–577 (2018). - PubMed
-
- Stary HC et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 92, 1355–1374 (1995). - PubMed
-
- Cochain C et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res 122, 1661–1674 (2018). - PubMed
Methods references
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL125863/HL/NHLBI NIH HHS/United States
- R35 HL144475/HL/NHLBI NIH HHS/United States
- R01 HL125224/HL/NHLBI NIH HHS/United States
- R35 GM133712/GM/NIGMS NIH HHS/United States
- R01 HL141425/HL/NHLBI NIH HHS/United States
- R01 HL148167/HL/NHLBI NIH HHS/United States
- R01 HL139478/HL/NHLBI NIH HHS/United States
- R01 HL164577/HL/NHLBI NIH HHS/United States
- R01 HL148239/HL/NHLBI NIH HHS/United States
- R01 HL135093/HL/NHLBI NIH HHS/United States
- R01 HL123370/HL/NHLBI NIH HHS/United States
- R01 HL130423/HL/NHLBI NIH HHS/United States
- R01 HL134817/HL/NHLBI NIH HHS/United States
- R00 HL125912/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
