

Widespread interspecific phylogenetic tree incongruence between mosquito-borne and insect-specific flaviviruses at hotspots originally identified in Zika virus
- PMID: 35591877
- PMCID: PMC9113262
- DOI: 10.1093/ve/veac027
Widespread interspecific phylogenetic tree incongruence between mosquito-borne and insect-specific flaviviruses at hotspots originally identified in Zika virus
Abstract
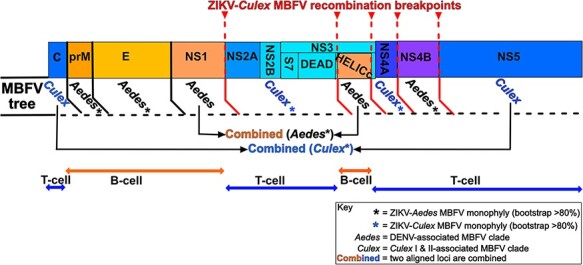
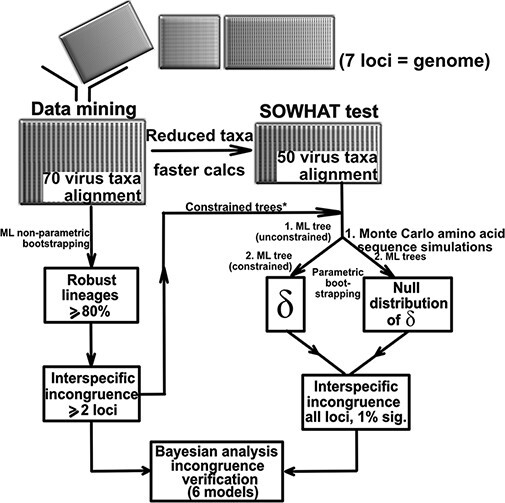
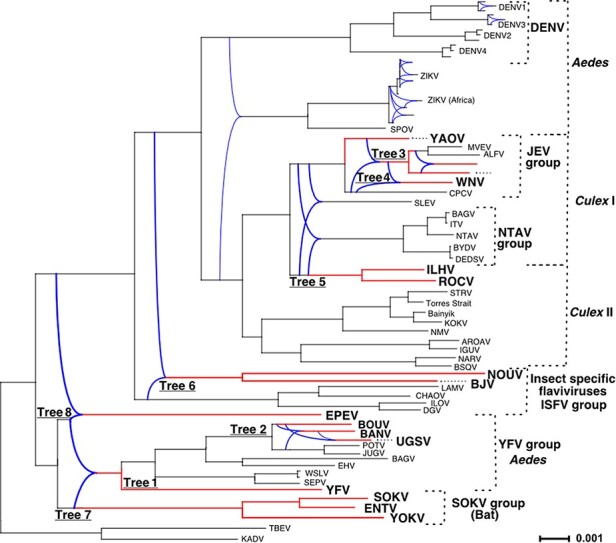
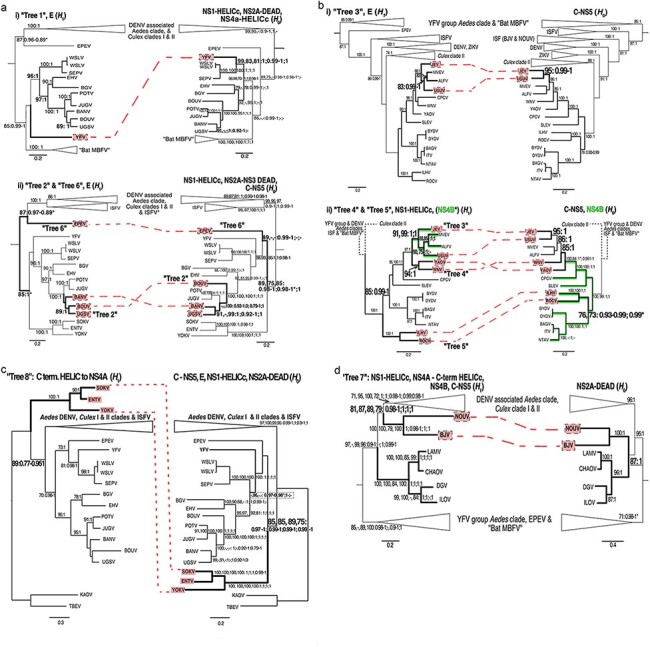
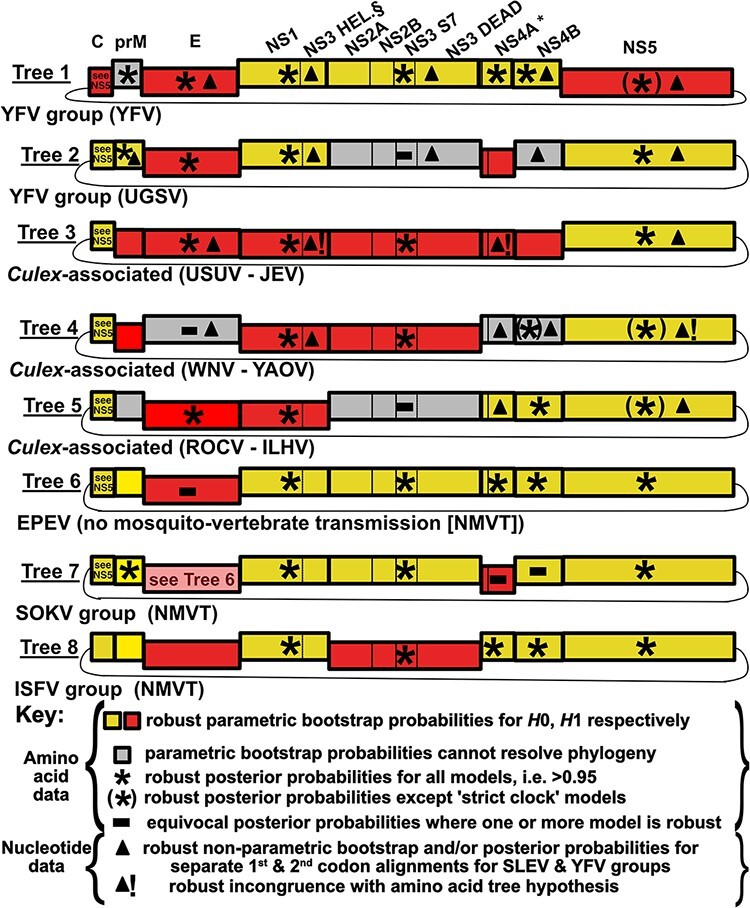
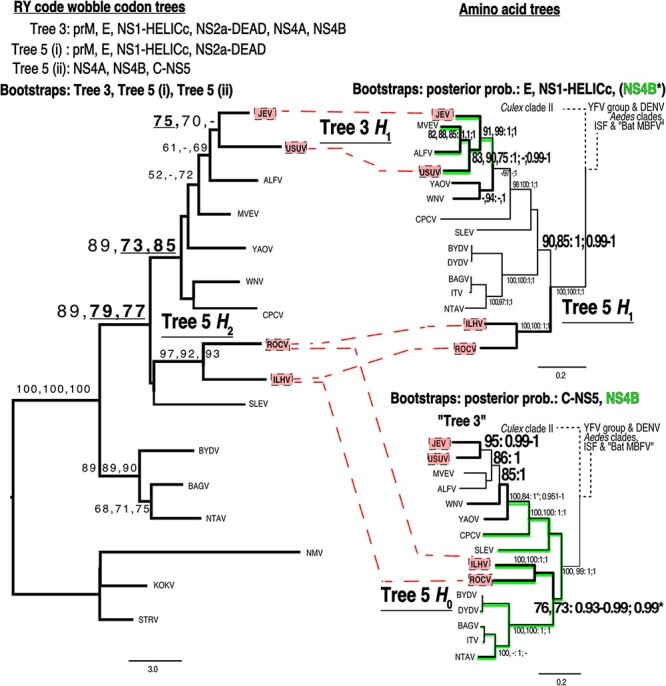
Intraspecies (homologous) phylogenetic incongruence, or 'tree conflict' between different loci within the same genome of mosquito-borne flaviviruses (MBFV), was first identified in dengue virus (DENV) and subsequently in Japanese encephalitis virus (JEV), St Louis encephalitis virus, and Zika virus (ZIKV). Recently, the first evidence of phylogenetic incongruence between interspecific members of the MBFV was reported in ZIKV and its close relative, Spondweni virus. Uniquely, these hybrid proteomes were derived from four incongruent trees involving an Aedes-associated DENV node (1 tree) and three different Culex-associated flavivirus nodes (3 trees). This analysis has now been extended across a wider spectrum of viruses within the MBFV lineage targeting the breakpoints between phylogenetic incongruent loci originally identified in ZIKV. Interspecies phylogenetic incongruence at these breakpoints was identified in 10 of 50 viruses within the MBFV lineage, representing emergent Aedes and Culex-associated viruses including JEV, West Nile virus, yellow fever virus, and insect-specific viruses. Thus, interspecies phylogenetic incongruence is widespread amongst the flaviviruses and is robustly associated with the specific breakpoints that coincide with the interspecific phylogenetic incongruence previously identified, inferring they are 'hotspots'. The incongruence amongst the emergent MBFV group was restricted to viruses within their respective associated epidemiological boundaries. This MBFV group was RY-coded at the third codon position ('wobble codon') to remove transition saturation. The resulting 'wobble codon' trees presented a single topology for the entire genome that lacked any robust evidence of phylogenetic incongruence between loci. Phylogenetic interspecific incongruence was therefore observed for exactly the same loci between amino acid and the RY-coded 'wobble codon' alignments and this incongruence represented either a major part, or the entire genomes. Maximum likelihood codon analysis revealed positive selection for the incongruent lineages. Positive selection could result in the same locus producing two opposing trees. These analyses for the clinically important MBFV suggest that robust interspecific phylogenetic incongruence resulted from amino acid selection. Convergent or parallel evolutions are evolutionary processes that would explain the observation, whilst interspecific recombination is unlikely.
Keywords: Mosquito-borne viruses; RY-coding; flavivirus; phylogenetic incongruence; recombination; selection.
© The Author(s) 2022. Published by Oxford University Press.
Figures
Similar articles
-
Recombination of B- and T-cell epitope-rich loci from Aedes- and Culex-borne flaviviruses shapes Zika virus epidemiology.Antiviral Res. 2020 Feb;174:104676. doi: 10.1016/j.antiviral.2019.104676. Epub 2019 Dec 16. Antiviral Res. 2020. PMID: 31837392
-
Detection of Quang Binh virus from mosquitoes in China.Virus Res. 2014 Feb 13;180:31-8. doi: 10.1016/j.virusres.2013.12.005. Epub 2013 Dec 14. Virus Res. 2014. PMID: 24342141
-
Novel flaviviruses from mosquitoes: mosquito-specific evolutionary lineages within the phylogenetic group of mosquito-borne flaviviruses.Virology. 2014 Sep;464-465:320-329. doi: 10.1016/j.virol.2014.07.015. Epub 2014 Aug 9. Virology. 2014. PMID: 25108382 Free PMC article.
-
Four human diseases with significant public health impact caused by mosquito-borne flaviviruses: West Nile, Zika, dengue and yellow fever.Semin Diagn Pathol. 2019 May;36(3):170-176. doi: 10.1053/j.semdp.2019.04.009. Epub 2019 Apr 17. Semin Diagn Pathol. 2019. PMID: 31006554 Review.
-
Dengue and other emerging flaviviruses.J Infect. 2001 Feb;42(2):104-15. doi: 10.1053/jinf.2001.0802. J Infect. 2001. PMID: 11531316 Review.
Cited by
-
Contrasting the Practices of Virus Isolation and Characterization between the Early Period in History and Modern Times: The Case of Japanese Encephalitis Virus.Viruses. 2022 Nov 26;14(12):2640. doi: 10.3390/v14122640. Viruses. 2022. PMID: 36560644 Free PMC article. Review.
-
Mechanisms of Yellow Fever Transmission: Gleaning the Overlooked Records of Importance and Identifying Problems, Puzzles, Serious Issues, Surprises and Research Questions.Viruses. 2024 Jan 4;16(1):84. doi: 10.3390/v16010084. Viruses. 2024. PMID: 38257784 Free PMC article. Review.
References
-
- Alfaro M. E. et al. (2003) ‘Bayes or Bootstrap? A Simulation Study Comparing the Performance of Bayesian Markov Chain Monte Carlo Sampling and Bootstrapping in Assessing Phylogenetic Confidence’, Molecular Biology and Evolution, 20: 255–66. - PubMed
-
- Bara J. J. et al. (2013) ‘Susceptibility of Larval Aedes Aegypti and Aedes Albopictus (Diptera: Culicidae) to Dengue Virus’, Journal of Medical Entomology, 50: 179–84. - PubMed
LinkOut - more resources
Full Text Sources
