

Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2
- PMID: 35594413
- PMCID: PMC9159519
- DOI: 10.1021/acs.chemrev.1c00965
Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2
Abstract
Despite tremendous efforts in the past two years, our understanding of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), virus-host interactions, immune response, virulence, transmission, and evolution is still very limited. This limitation calls for further in-depth investigation. Computational studies have become an indispensable component in combating coronavirus disease 2019 (COVID-19) due to their low cost, their efficiency, and the fact that they are free from safety and ethical constraints. Additionally, the mechanism that governs the global evolution and transmission of SARS-CoV-2 cannot be revealed from individual experiments and was discovered by integrating genotyping of massive viral sequences, biophysical modeling of protein-protein interactions, deep mutational data, deep learning, and advanced mathematics. There exists a tsunami of literature on the molecular modeling, simulations, and predictions of SARS-CoV-2 and related developments of drugs, vaccines, antibodies, and diagnostics. To provide readers with a quick update about this literature, we present a comprehensive and systematic methodology-centered review. Aspects such as molecular biophysics, bioinformatics, cheminformatics, machine learning, and mathematics are discussed. This review will be beneficial to researchers who are looking for ways to contribute to SARS-CoV-2 studies and those who are interested in the status of the field.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
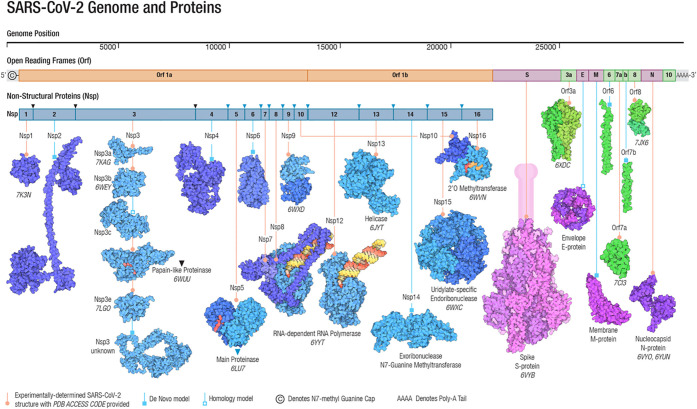
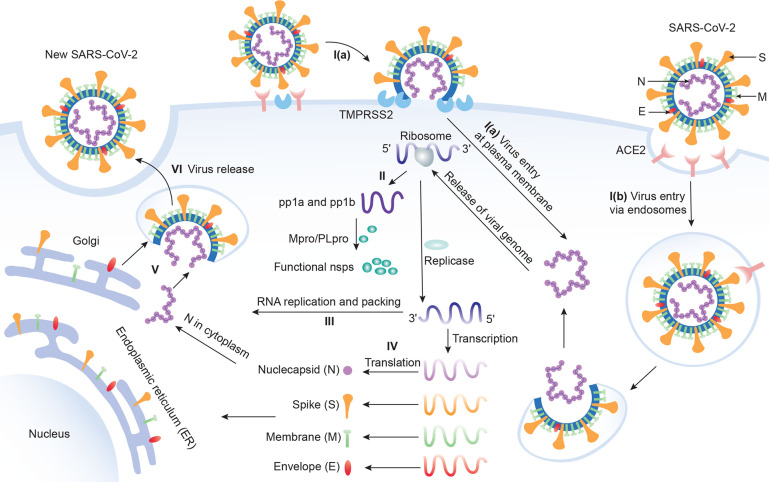
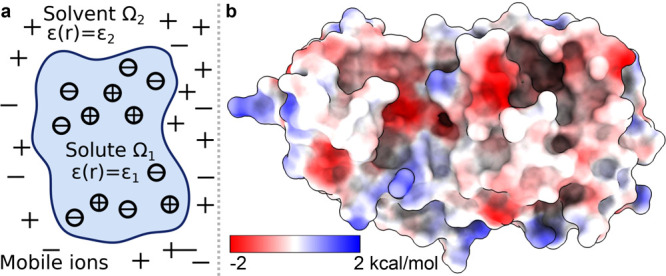
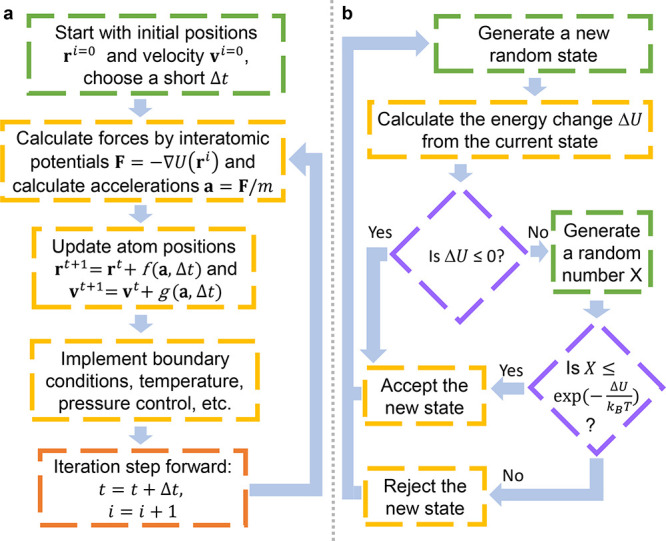
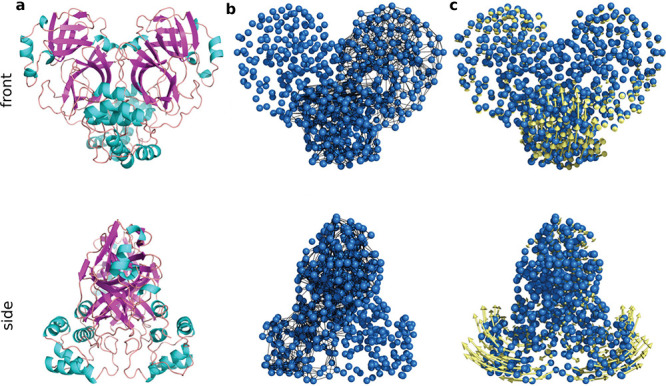
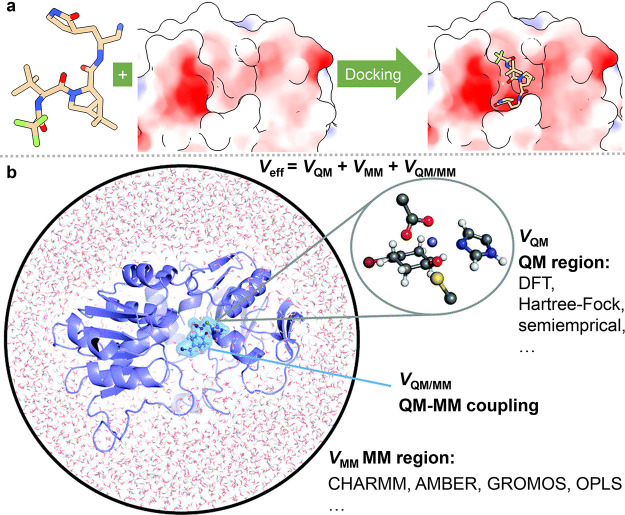
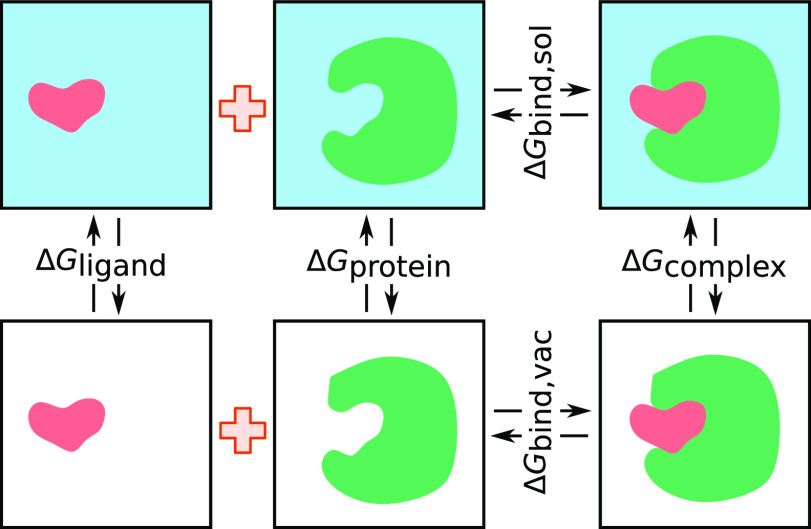
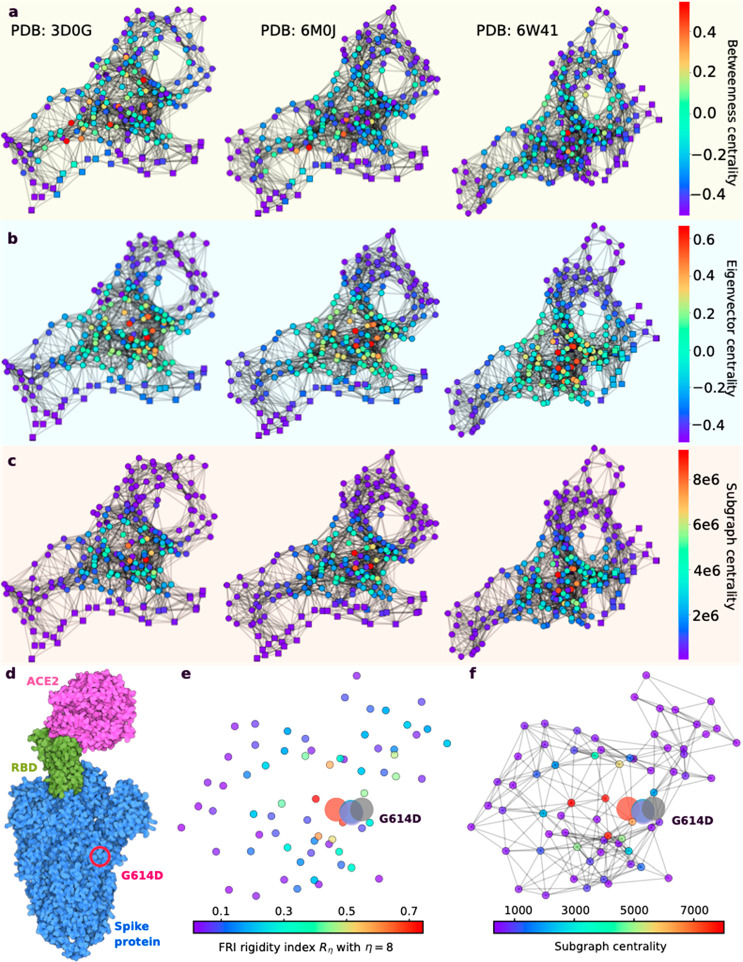

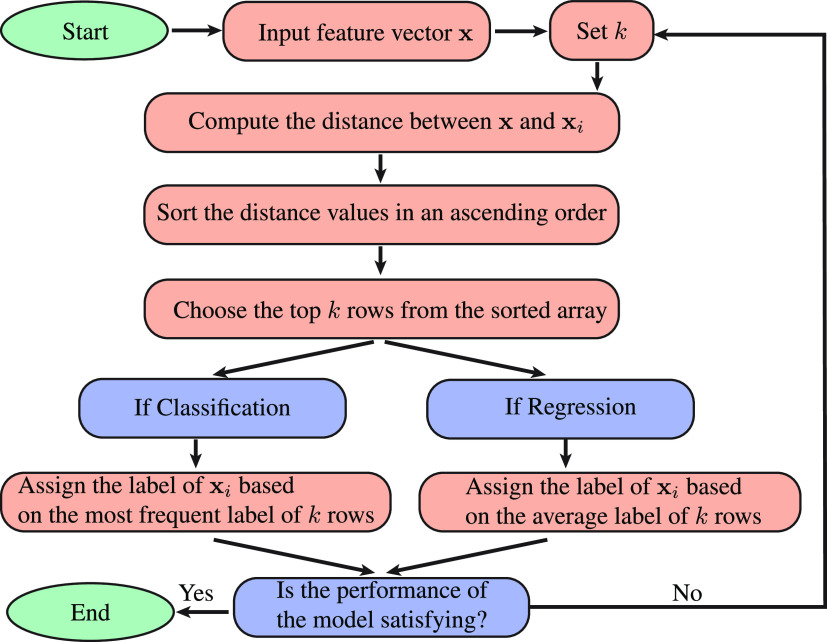
 , k shows the number of
nearest neighbors, and
, k shows the number of
nearest neighbors, and  is a feature representation of the training
set.
is a feature representation of the training
set.
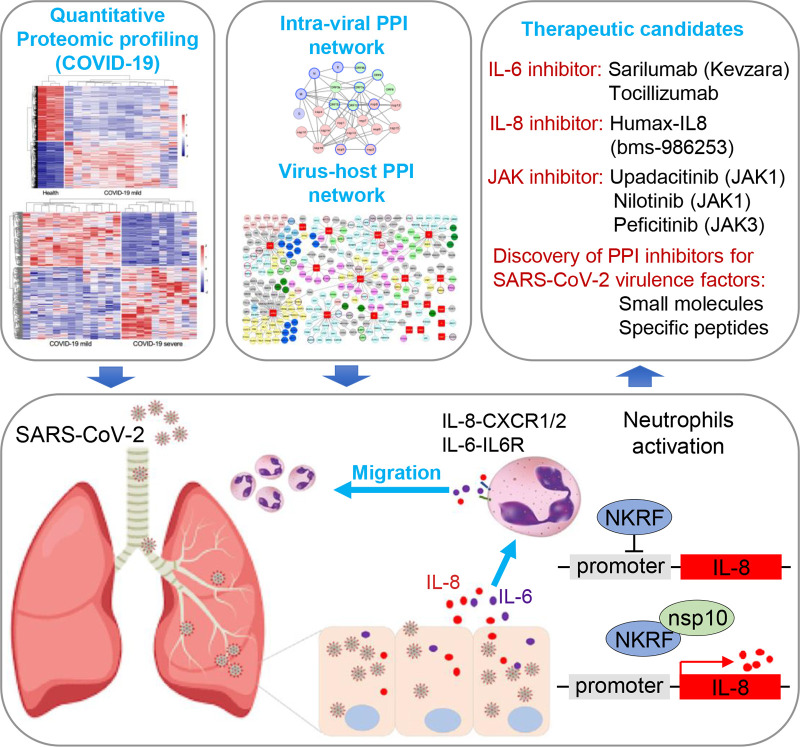
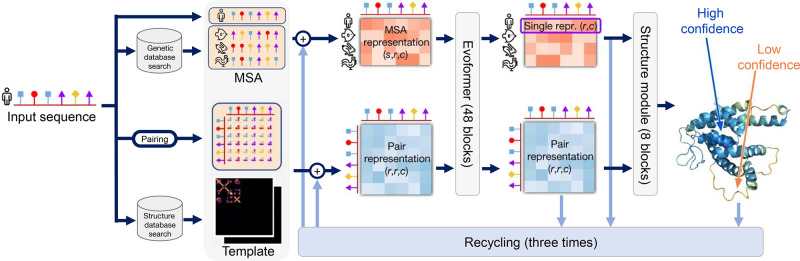
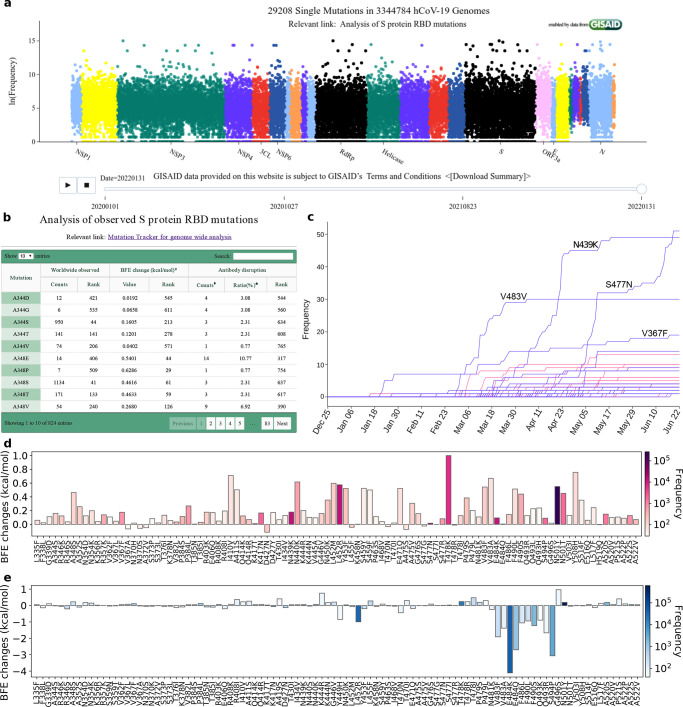
Similar articles
-
Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area.mBio. 2020 Oct 30;11(6):e02707-20. doi: 10.1128/mBio.02707-20. mBio. 2020. PMID: 33127862 Free PMC article.
-
Molecular Insights of SARS-CoV-2 Infection and Molecular Treatments.Curr Mol Med. 2022;22(7):621-639. doi: 10.2174/1566524021666211013121831. Curr Mol Med. 2022. PMID: 34645374 Review.
-
Comprehensive analyses of bioinformatics applications in the fight against COVID-19 pandemic.Comput Biol Chem. 2021 Dec;95:107599. doi: 10.1016/j.compbiolchem.2021.107599. Epub 2021 Nov 2. Comput Biol Chem. 2021. PMID: 34773807 Free PMC article. Review.
-
Biological, clinical and epidemiological features of COVID-19, SARS and MERS and AutoDock simulation of ACE2.Infect Dis Poverty. 2020 Jul 20;9(1):99. doi: 10.1186/s40249-020-00691-6. Infect Dis Poverty. 2020. PMID: 32690096 Free PMC article. Review.
-
The British variant of the new coronavirus-19 (Sars-Cov-2) should not create a vaccine problem.J Biol Regul Homeost Agents. 2021 Jan-Feb;35(1):1-4. doi: 10.23812/21-3-E. J Biol Regul Homeost Agents. 2021. PMID: 33377359
Cited by
-
Conformational Stability and Denaturation Processes of Proteins Investigated by Electrophoresis under Extreme Conditions.Molecules. 2022 Oct 13;27(20):6861. doi: 10.3390/molecules27206861. Molecules. 2022. PMID: 36296453 Free PMC article. Review.
-
Assessing the Potential Contribution of In Silico Studies in Discovering Drug Candidates That Interact with Various SARS-CoV-2 Receptors.Int J Mol Sci. 2023 Oct 24;24(21):15518. doi: 10.3390/ijms242115518. Int J Mol Sci. 2023. PMID: 37958503 Free PMC article. Review.
-
An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor.Molecules. 2021 Apr 20;26(8):2383. doi: 10.3390/molecules26082383. Molecules. 2021. PMID: 33923909 Free PMC article.
-
A Novel 3-Gene Signature for Identifying COVID-19 Patients Based on Bioinformatics and Machine Learning.Genes (Basel). 2022 Sep 8;13(9):1602. doi: 10.3390/genes13091602. Genes (Basel). 2022. PMID: 36140771 Free PMC article.
-
Biophysics of SARS-CoV-2 spike protein's receptor-binding domain interaction with ACE2 and neutralizing antibodies: from computation to functional insights.Biophys Rev. 2025 Mar 8;17(2):309-333. doi: 10.1007/s12551-025-01276-z. eCollection 2025 Apr. Biophys Rev. 2025. PMID: 40376405 Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
