

Compatibility rules of human enhancer and promoter sequences
- PMID: 35594906
- PMCID: PMC9262863
- DOI: 10.1038/s41586-022-04877-w
Compatibility rules of human enhancer and promoter sequences
Abstract
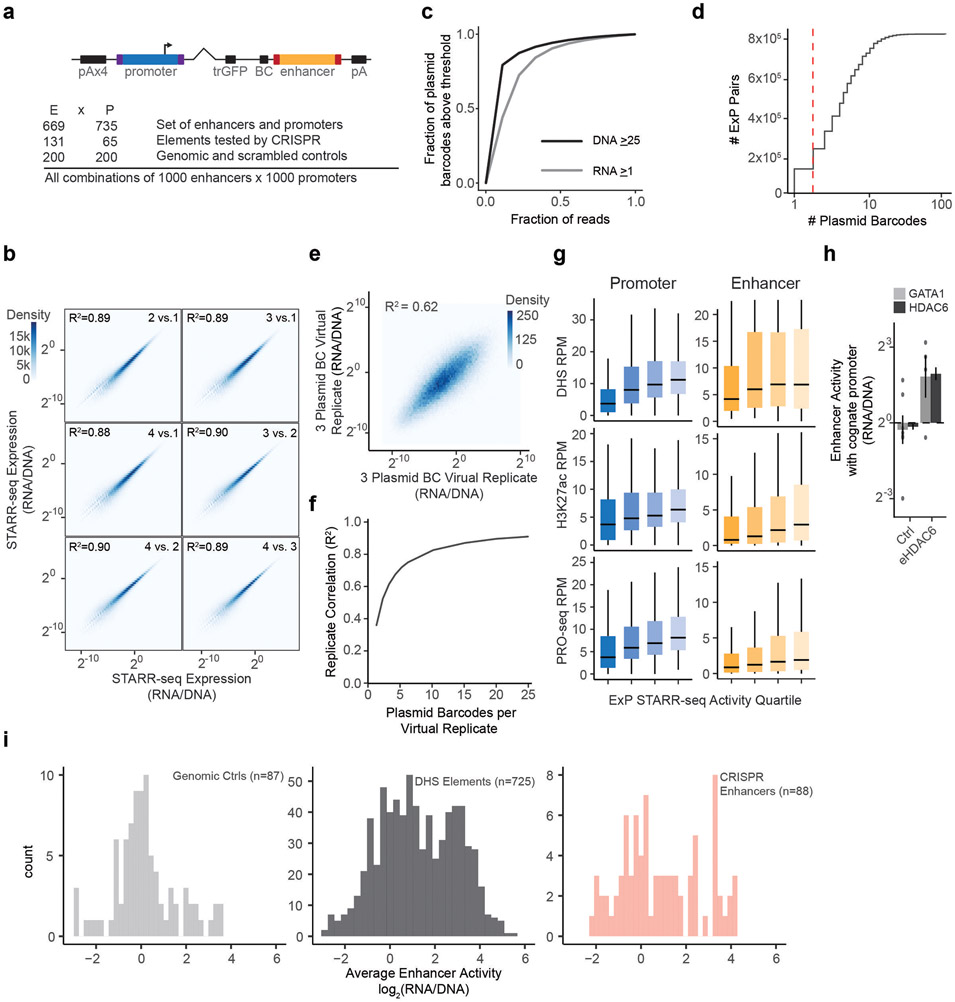
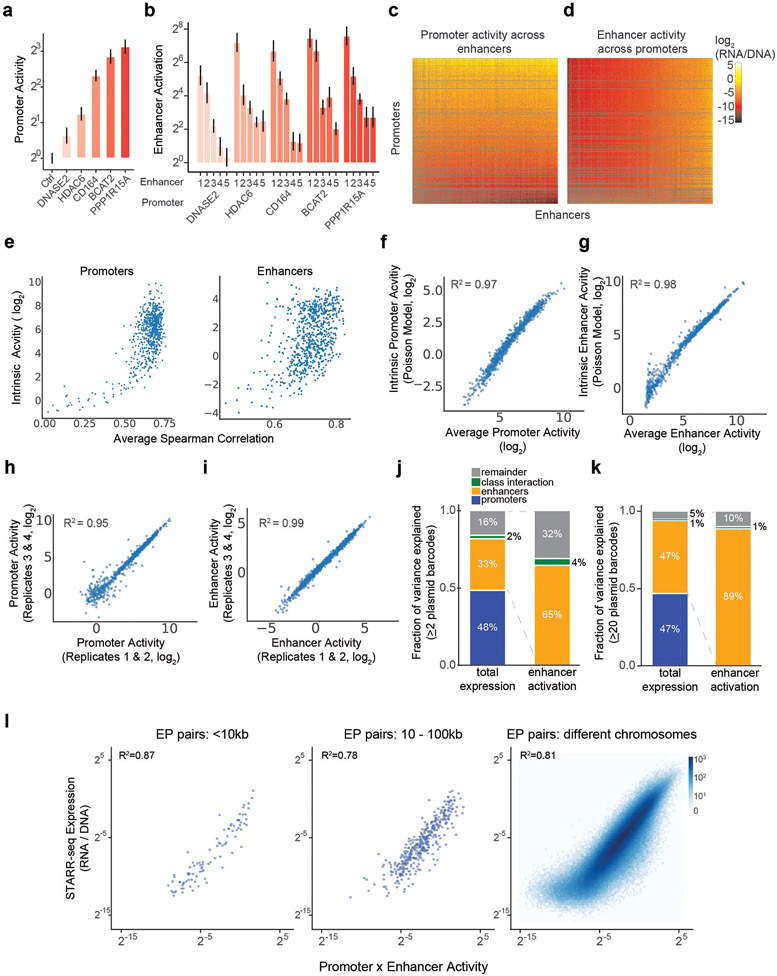
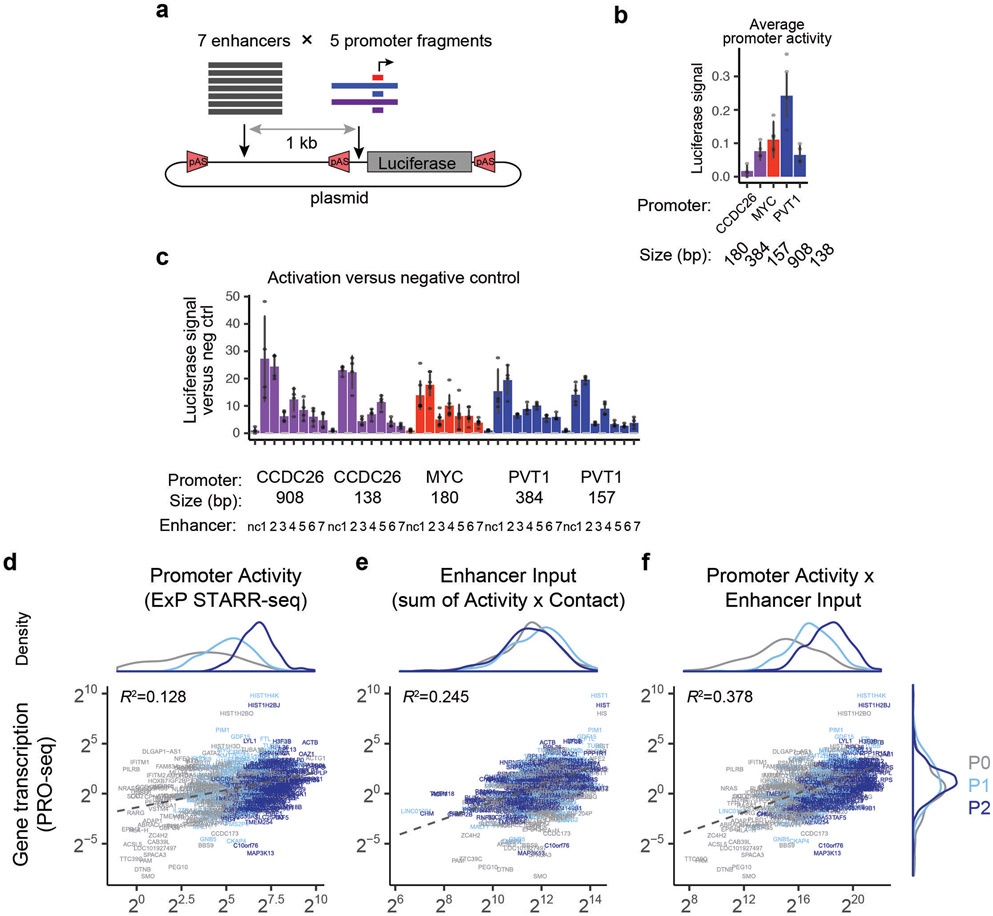
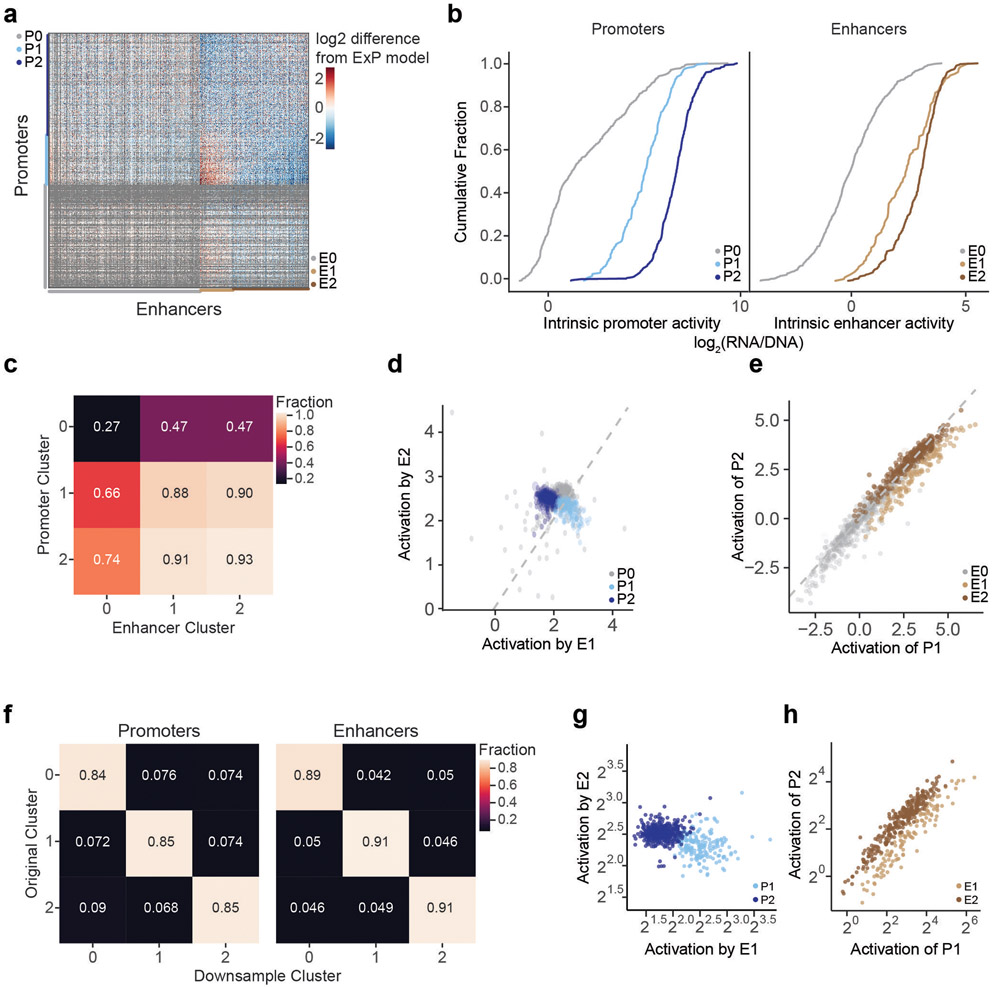
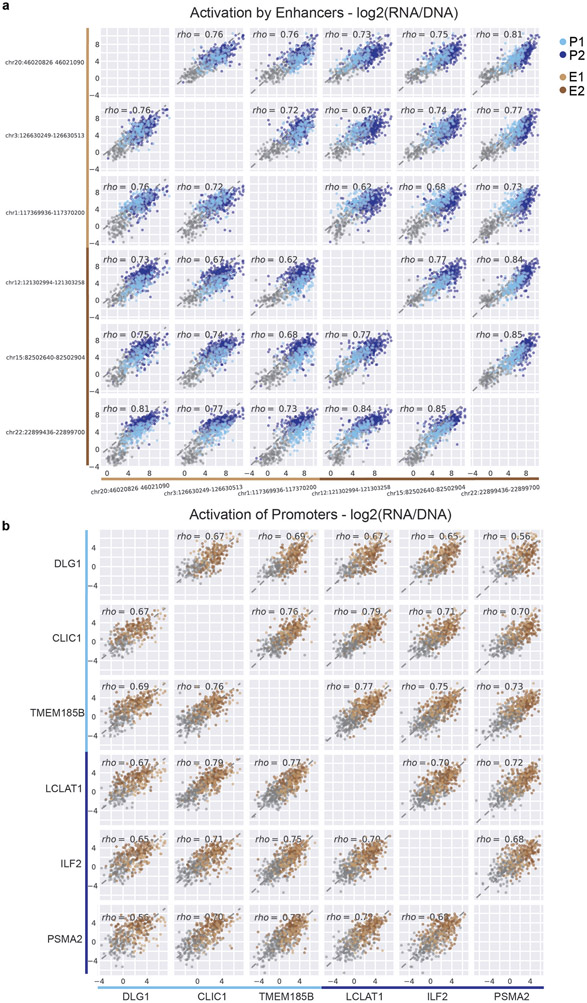
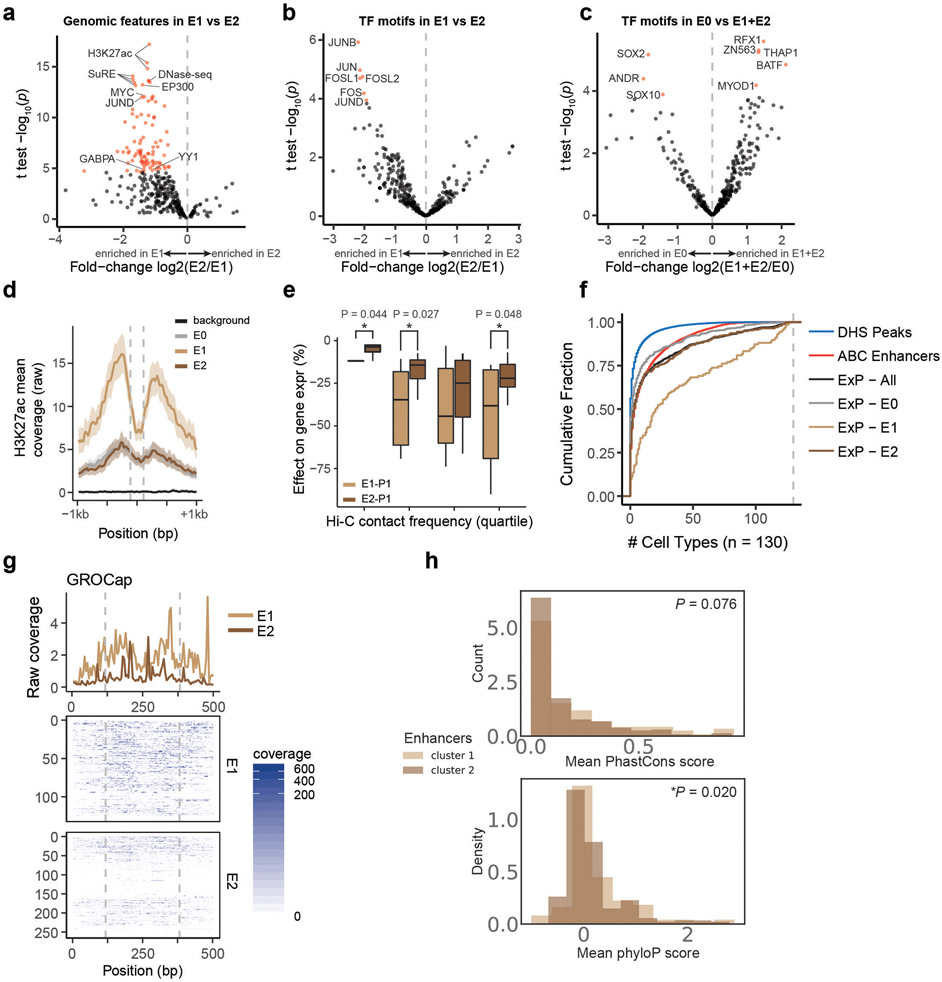
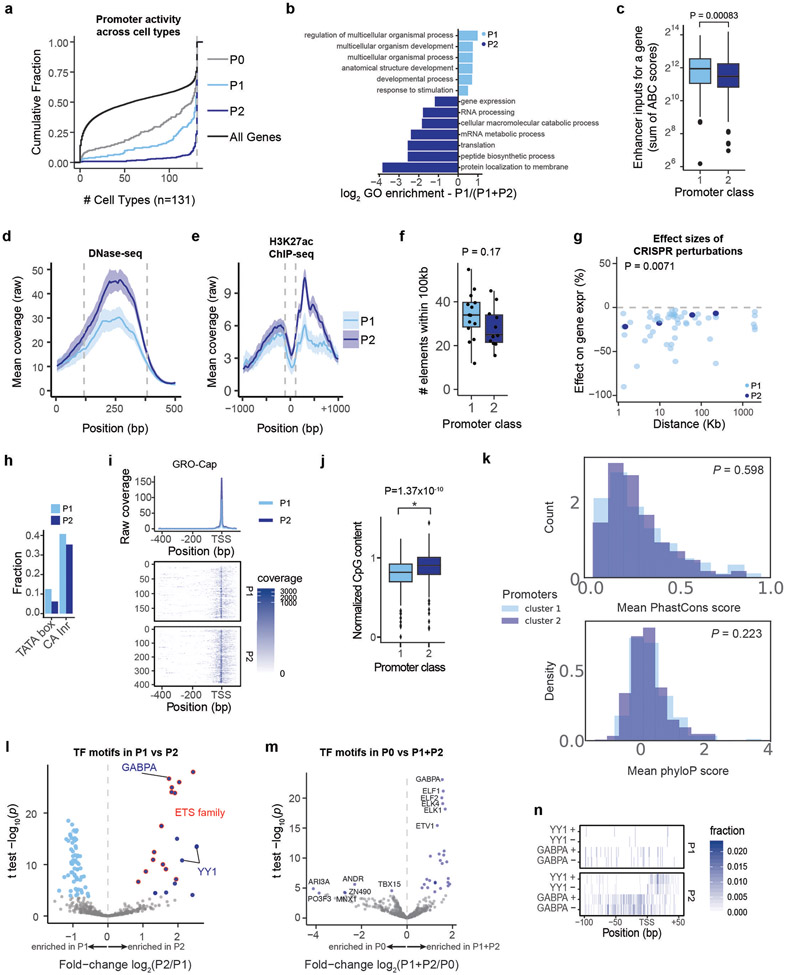
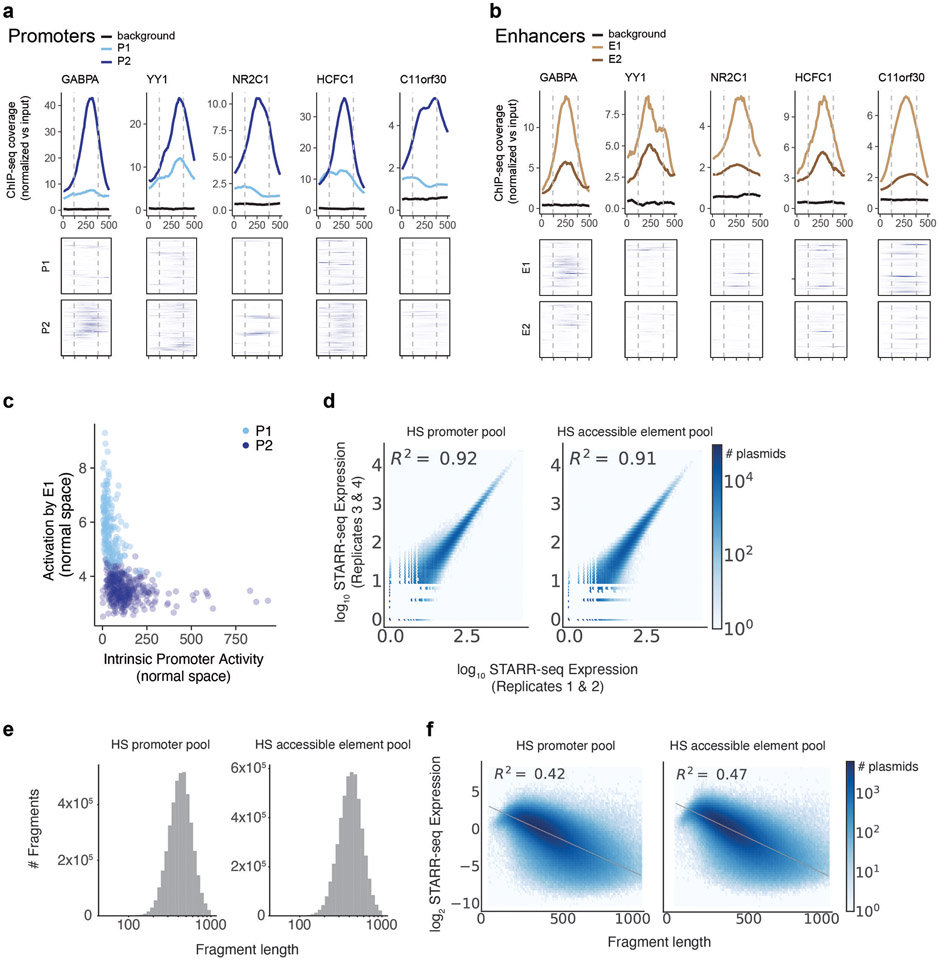
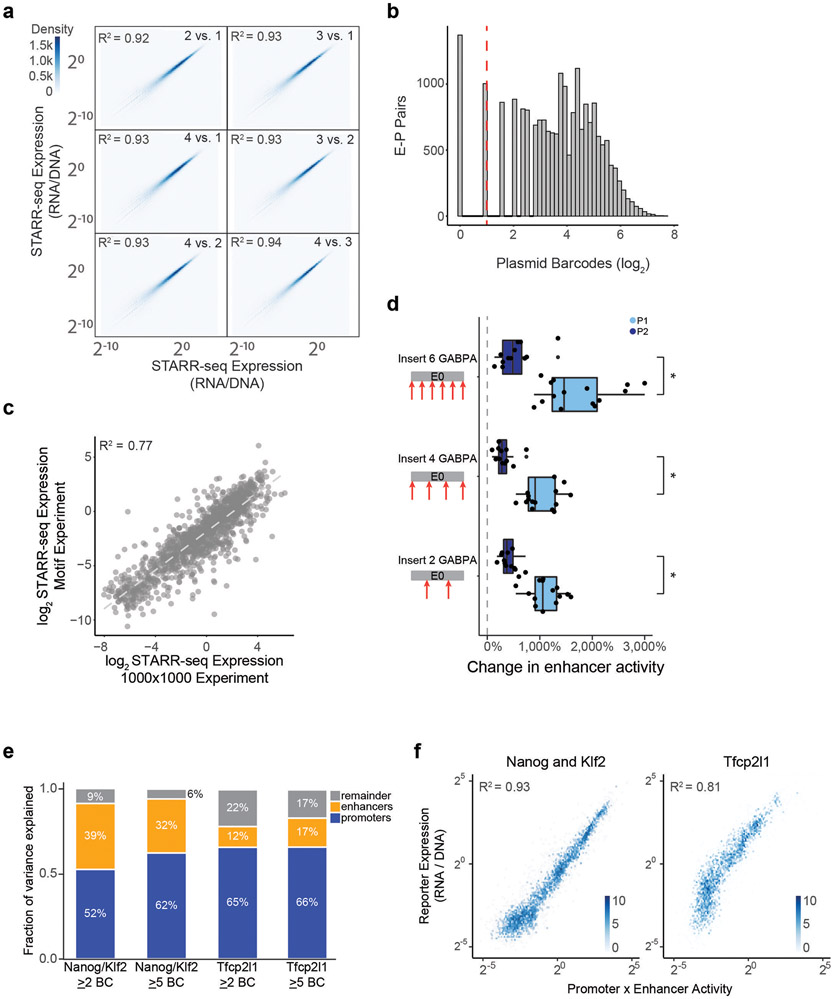
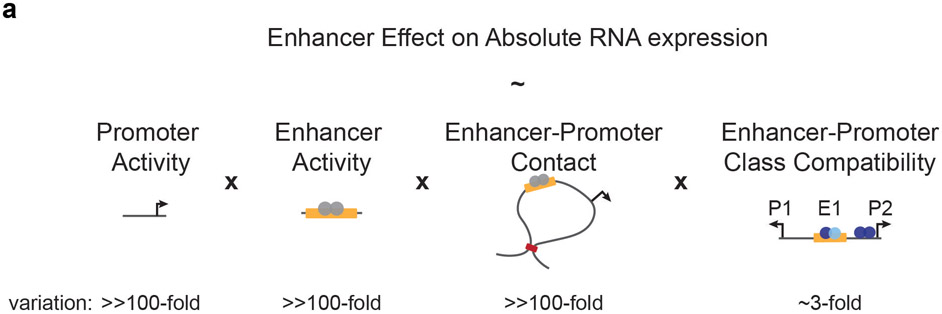
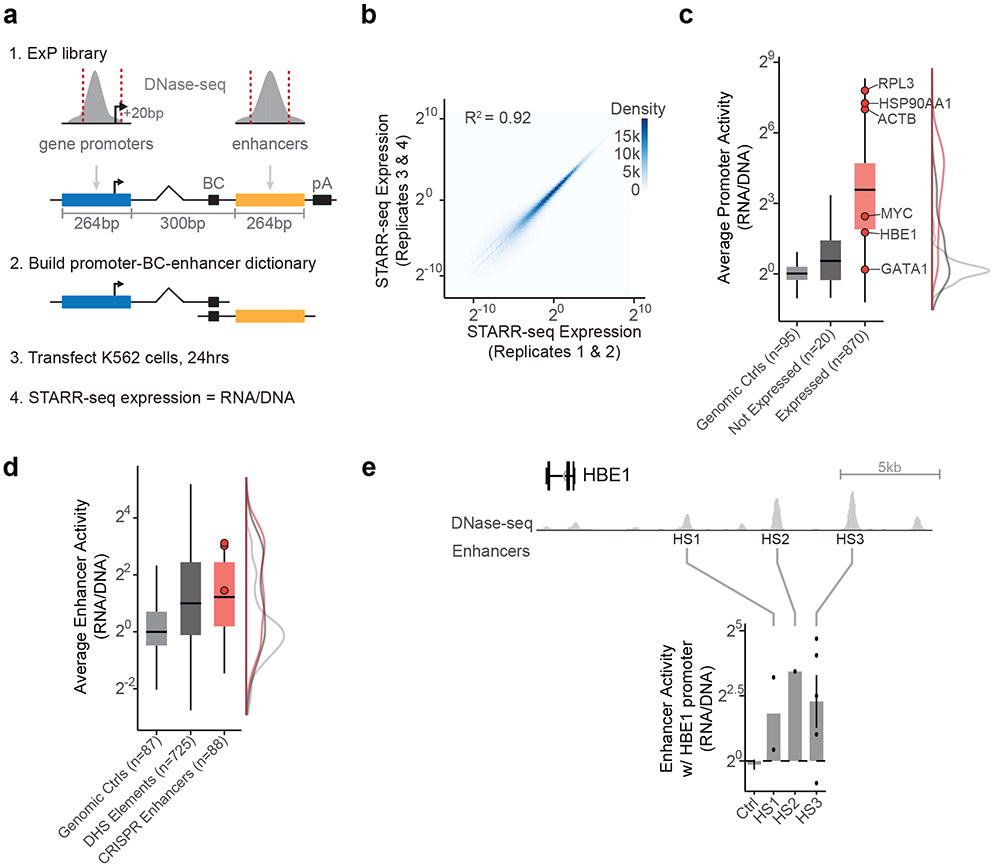
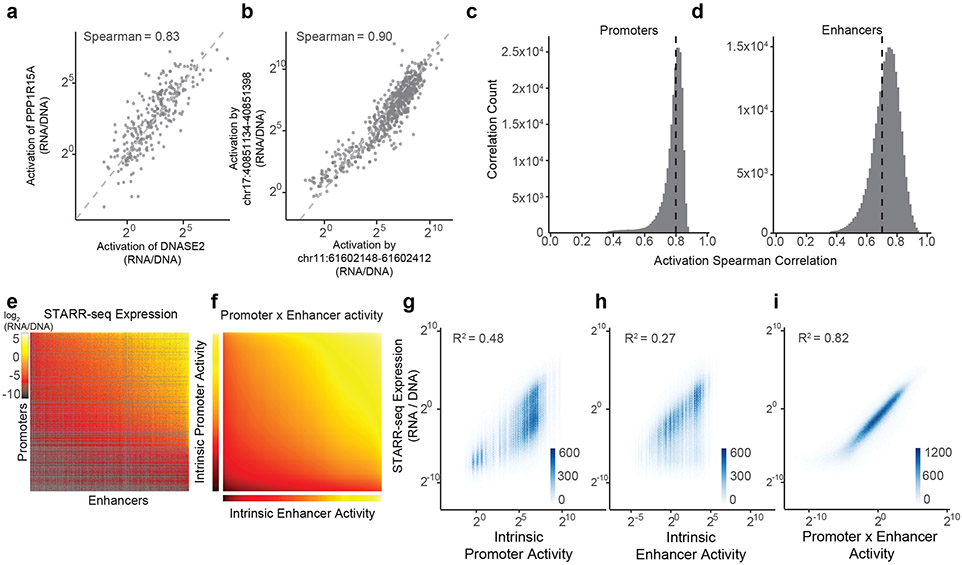
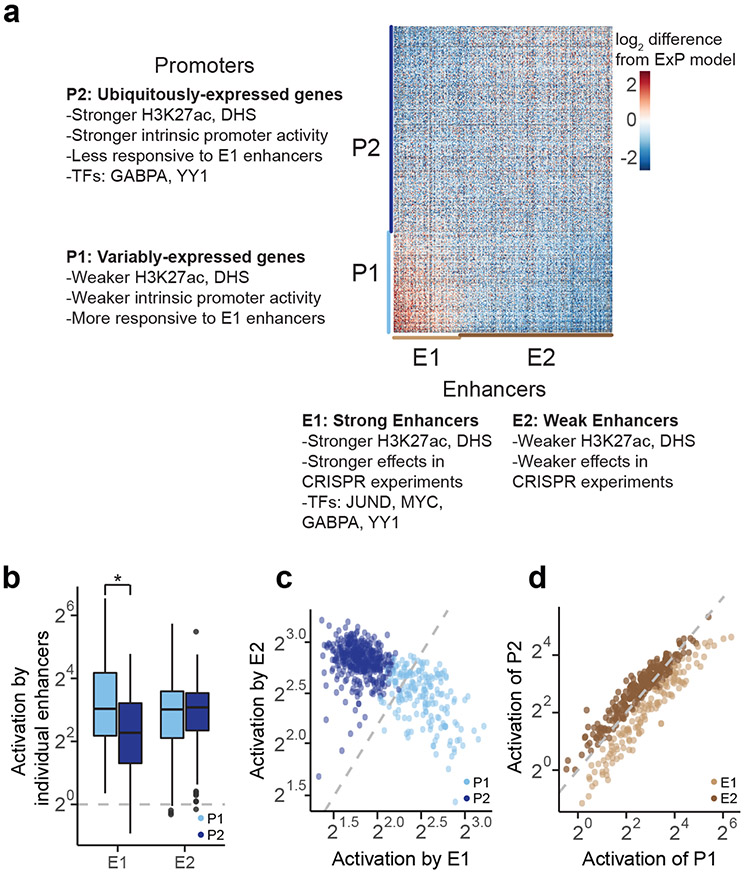
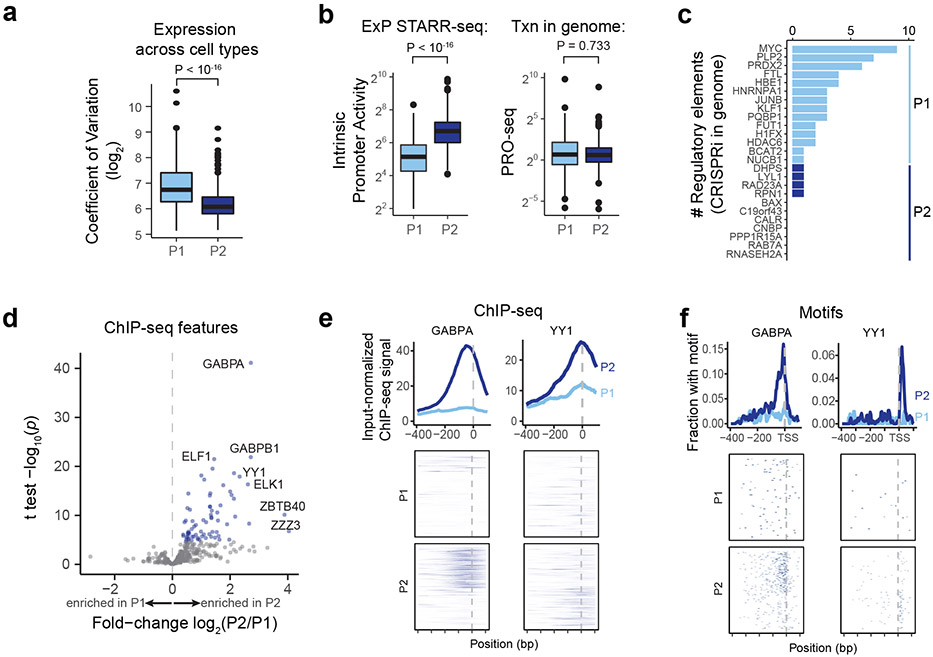
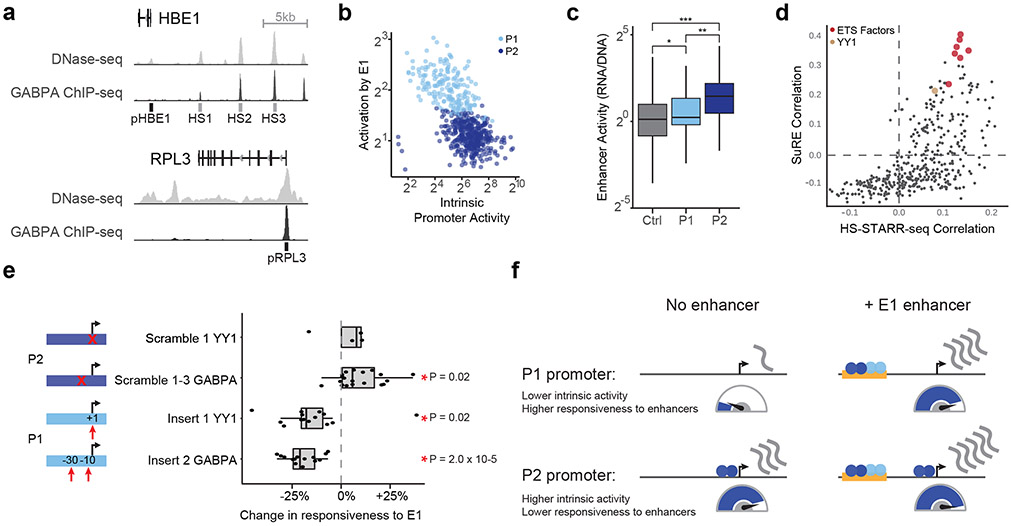
Gene regulation in the human genome is controlled by distal enhancers that activate specific nearby promoters1. A proposed model for this specificity is that promoters have sequence-encoded preferences for certain enhancers, for example, mediated by interacting sets of transcription factors or cofactors2. This 'biochemical compatibility' model has been supported by observations at individual human promoters and by genome-wide measurements in Drosophila3-9. However, the degree to which human enhancers and promoters are intrinsically compatible has not yet been systematically measured, and how their activities combine to control RNA expression remains unclear. Here we design a high-throughput reporter assay called enhancer × promoter self-transcribing active regulatory region sequencing (ExP STARR-seq) and applied it to examine the combinatorial compatibilities of 1,000 enhancer and 1,000 promoter sequences in human K562 cells. We identify simple rules for enhancer-promoter compatibility, whereby most enhancers activate all promoters by similar amounts, and intrinsic enhancer and promoter activities multiplicatively combine to determine RNA output (R2 = 0.82). In addition, two classes of enhancers and promoters show subtle preferential effects. Promoters of housekeeping genes contain built-in activating motifs for factors such as GABPA and YY1, which decrease the responsiveness of promoters to distal enhancers. Promoters of variably expressed genes lack these motifs and show stronger responsiveness to enhancers. Together, this systematic assessment of enhancer-promoter compatibility suggests a multiplicative model tuned by enhancer and promoter class to control gene transcription in the human genome.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
Comment in
-
The Cupid shuffle: Do enhancers prefer specific promoters?Mol Cell. 2022 Jul 7;82(13):2357-2359. doi: 10.1016/j.molcel.2022.06.014. Mol Cell. 2022. PMID: 35803216 Free PMC article.
References
Main Text References
Additional References
-
- Anscombe FJ THE TRANSFORMATION OF POISSON, BINOMIAL AND NEGATIVE-BINOMIAL DATA. Biometrika vol. 35 246–254 (1948).
-
- Vanhille L et al. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun 6, 6905 (2015). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
