

Pirtobrutinib inhibits wild-type and mutant Bruton's tyrosine kinase-mediated signaling in chronic lymphocytic leukemia
- PMID: 35595730
- PMCID: PMC9123190
- DOI: 10.1038/s41408-022-00675-9
Pirtobrutinib inhibits wild-type and mutant Bruton's tyrosine kinase-mediated signaling in chronic lymphocytic leukemia
Abstract
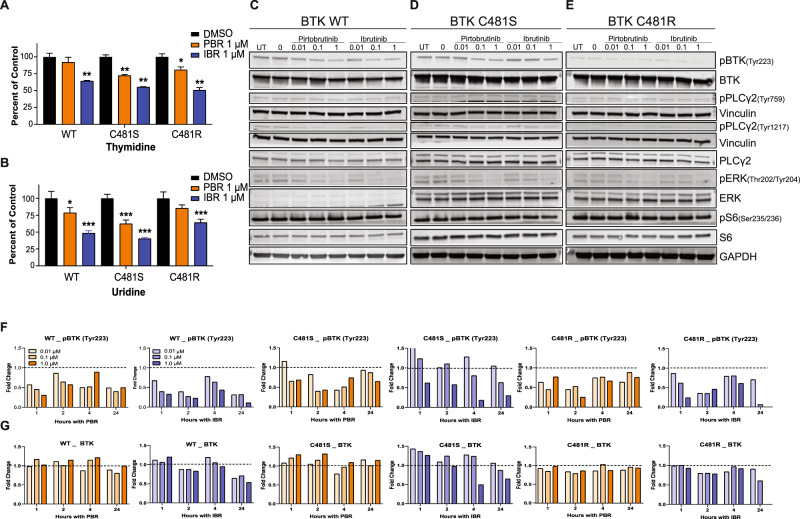
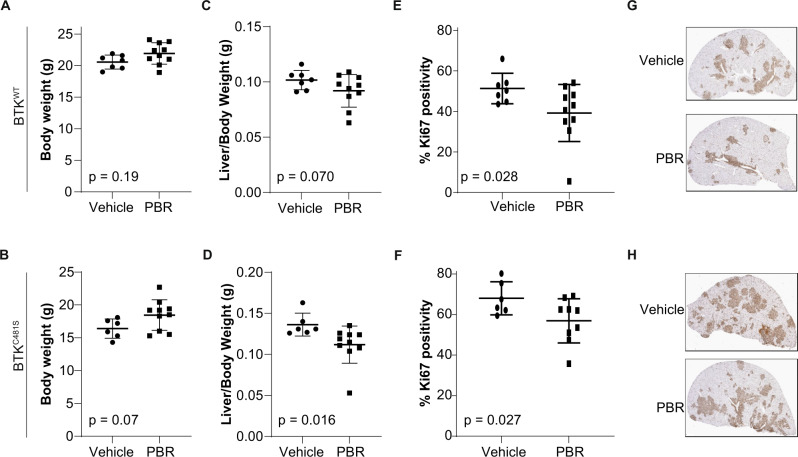
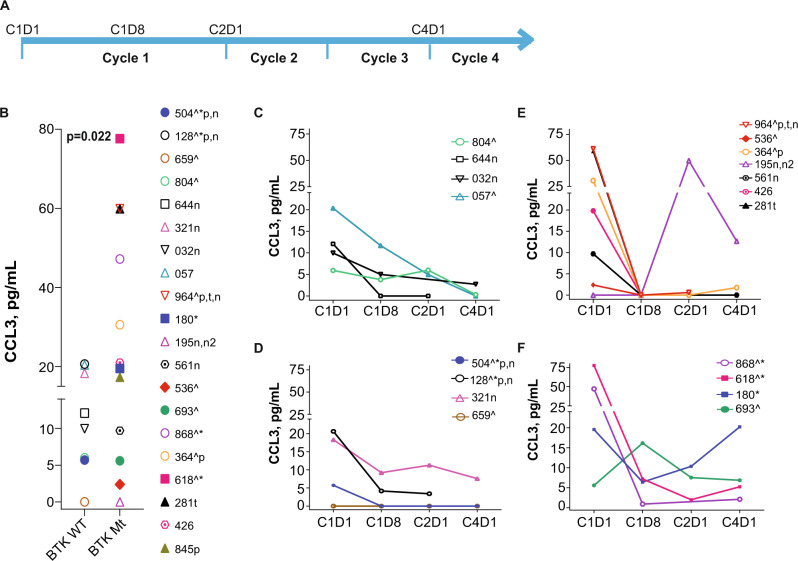
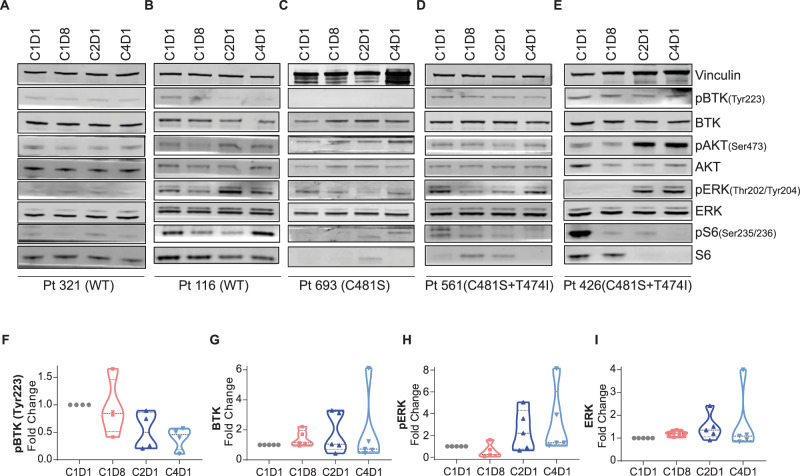
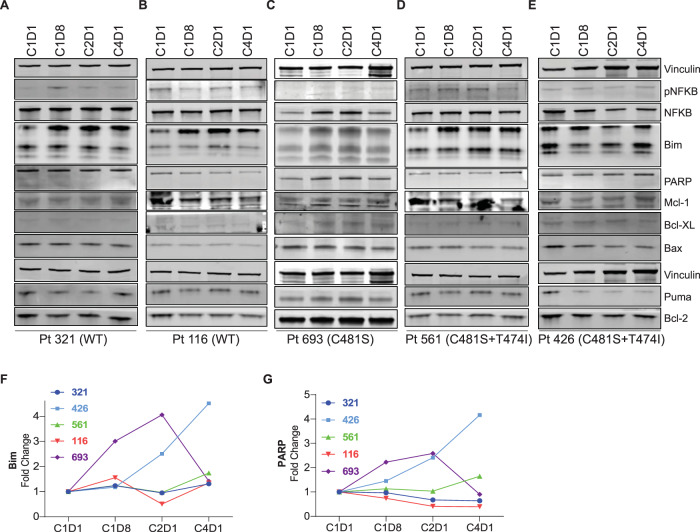
Pirtobrutinib (LOXO-305), a reversible inhibitor of Bruton's tyrosine kinase (BTK), was designed as an alternative strategy to treat ibrutinib-resistant disease that develops due to C481 kinase domain mutations. The clinical activity of pirtobrutinib has been demonstrated in CLL, but the mechanism of action has not been investigated. We evaluated pirtobrutinib in 4 model systems: first, MEC-1, a CLL cell line overexpressing BTKWT, BTKC481S, or BTKC481R; second, murine models driven by MEC-1 overexpressing BTKWT or BTKC481S; third, in vitro incubations of primary CLL cells; and finally, CLL patients during pirtobrutinib therapy (NCT03740529, ClinicalTrials.gov). Pirtobrutinib inhibited BTK activation as well as downstream signaling in MEC-1 isogenic cells overexpressing BTKWT, BTKC481S, or BTKC481R. In mice, overall survival was short due to aggressive disease. Pirtobrutinib treatment for 2 weeks led to reduction of spleen and liver weight in BTKWT and BTKC481S cells, respectively. In vitro incubations of CLL cells harboring wild-type or mutant BTK had inhibition of the BCR pathway with either ibrutinib or pirtobrutinib treatment. Pirtobrutinib therapy resulted in inhibition of BTK phosphorylation and downstream signaling initially in all cases irrespective of their BTK profile, but these effects started to revert in cases with other BCR pathway mutations such as PLCG2 or PLEKHG5. Levels of CCL3 and CCL4 in plasma were marginally higher in patients with mutated BTK; however, there was a bimodal distribution. Both chemokines were decreased at early time points and mimicked BCR pathway protein changes. Collectively, these results demonstrate that pirtobrutinib is an effective BTK inhibitor for CLL harboring wild-type or mutant BTK as observed by changes in CCL3 and CCL4 biomarkers and suggest that alterations in BCR pathway signaling are the mechanism for its clinical effects. Long-term evaluation is needed for BTK gatekeeper residue variation along with pathologic kinase substitution or mutations in other proteins in the BCR pathway.
© 2022. The Author(s).
Conflict of interest statement
Specifically, for the current investigations, VG received sponsored research agreement from Loxo Oncology (now a subsidiary of Lilly Oncology). Previously, for other investigations, VG received research funding from Pharmacyclics, Acerta, Gilead Sciences, Sunesis, Infinity, AbbVie, and Clear Creek Bio. WGW received research funding from GSK/Novartis, AbbVie, Genentech, Pharmacyclics, AstraZeneca/Acerta Pharma, Gilead Sciences, Juno Therapeutics, KITE Pharma, Sunesis, Miragen, Oncternal Therapeutics, Cyclacel, Loxo Oncology, Janssen, and Xencor. The remaining authors declare no competing financial interests.
Figures


Similar articles
-
Transcriptomic and proteomic differences in BTK-WT and BTK-mutated CLL and their changes during therapy with pirtobrutinib.Blood Adv. 2024 Sep 10;8(17):4487-4501. doi: 10.1182/bloodadvances.2023012360. Blood Adv. 2024. PMID: 38968154 Free PMC article.
-
Pirtobrutinib targets BTK C481S in ibrutinib-resistant CLL but second-site BTK mutations lead to resistance.Blood Adv. 2023 May 9;7(9):1929-1943. doi: 10.1182/bloodadvances.2022008447. Blood Adv. 2023. PMID: 36287227 Free PMC article.
-
Noncatalytic Bruton's tyrosine kinase activates PLCγ2 variants mediating ibrutinib resistance in human chronic lymphocytic leukemia cells.J Biol Chem. 2020 Apr 24;295(17):5717-5736. doi: 10.1074/jbc.RA119.011946. Epub 2020 Mar 17. J Biol Chem. 2020. PMID: 32184360 Free PMC article.
-
A Review of Resistance Mechanisms to Bruton's Kinase Inhibitors in Chronic Lymphocytic Leukemia.Int J Mol Sci. 2024 May 11;25(10):5246. doi: 10.3390/ijms25105246. Int J Mol Sci. 2024. PMID: 38791284 Free PMC article. Review.
-
Resistance mechanisms and approach to chronic lymphocytic leukemia after BTK inhibitor therapy.Leuk Lymphoma. 2025 Jul;66(7):1176-1188. doi: 10.1080/10428194.2025.2466101. Epub 2025 Feb 19. Leuk Lymphoma. 2025. PMID: 39972943 Review.
Cited by
-
Pharmacological profiling in CLL patients during pirtobrutinib therapy and disease progression.Res Sq [Preprint]. 2025 Mar 31:rs.3.rs-6249480. doi: 10.21203/rs.3.rs-6249480/v1. Res Sq. 2025. PMID: 40235506 Free PMC article. Preprint.
-
Pyrazole: an emerging privileged scaffold in drug discovery.Future Med Chem. 2023 Nov;15(21):2011-2023. doi: 10.4155/fmc-2023-0207. Epub 2023 Nov 7. Future Med Chem. 2023. PMID: 37933613 Free PMC article. Review.
-
Transcriptomic and proteomic differences in BTK-WT and BTK-mutated CLL and their changes during therapy with pirtobrutinib.Blood Adv. 2024 Sep 10;8(17):4487-4501. doi: 10.1182/bloodadvances.2023012360. Blood Adv. 2024. PMID: 38968154 Free PMC article.
-
B cell receptor signaling and associated pathways in the pathogenesis of chronic lymphocytic leukemia.Front Oncol. 2024 Feb 26;14:1339620. doi: 10.3389/fonc.2024.1339620. eCollection 2024. Front Oncol. 2024. PMID: 38469232 Free PMC article. Review.
-
Optimal use of BTK inhibitors in Waldenström's macroglobulinemia: combination or single drug approach?Ther Adv Hematol. 2024 Dec 23;15:20406207241308771. doi: 10.1177/20406207241308771. eCollection 2024. Ther Adv Hematol. 2024. PMID: 39734591 Free PMC article. Review.
References
-
- Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia blood. Blood J Am Soc Hematol. 2011;118:4313–20. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
