

Automated next-generation profiling of genomic alterations in human cancers
- PMID: 35595835
- PMCID: PMC9123004
- DOI: 10.1038/s41467-022-30380-x
Automated next-generation profiling of genomic alterations in human cancers
Abstract
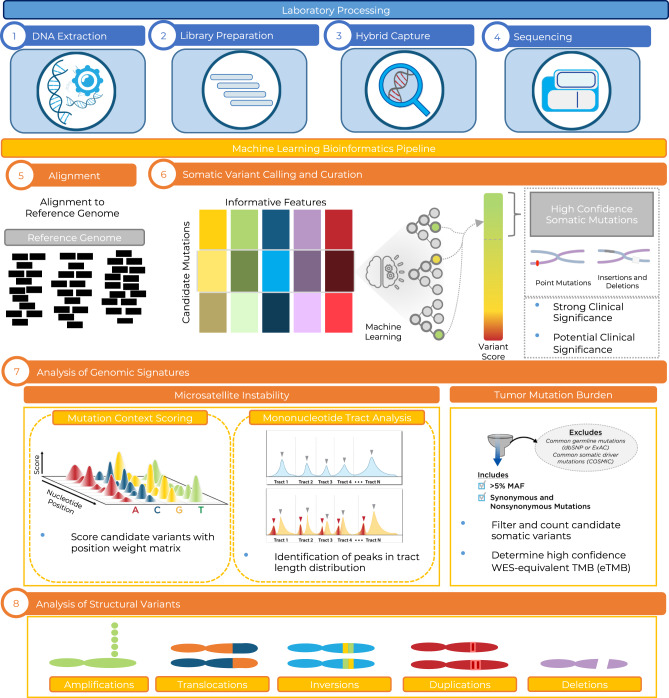
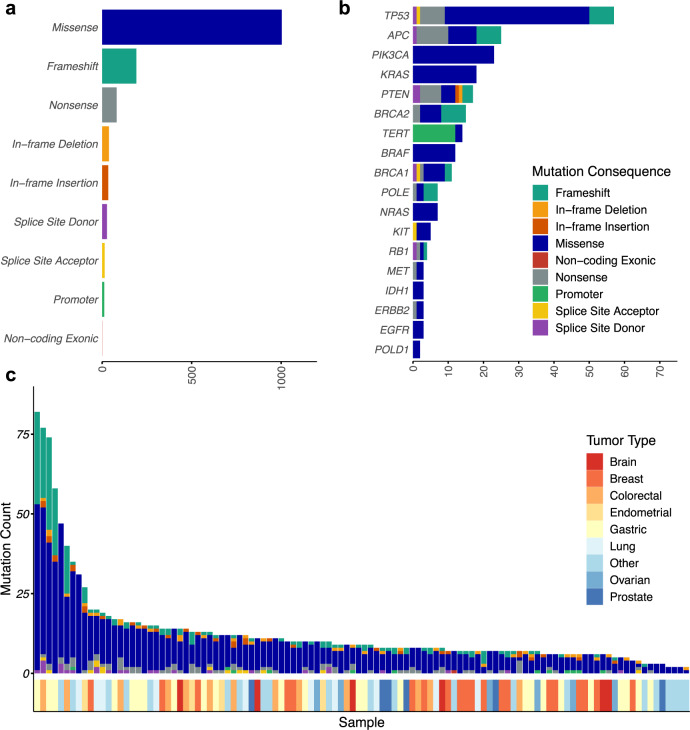
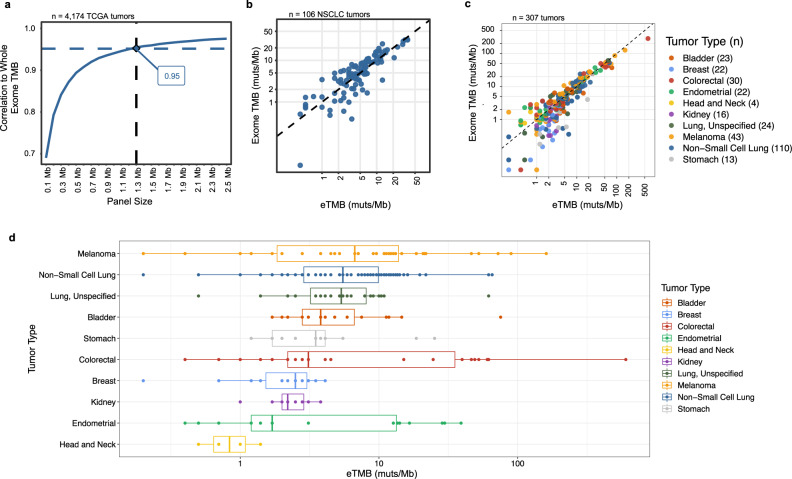
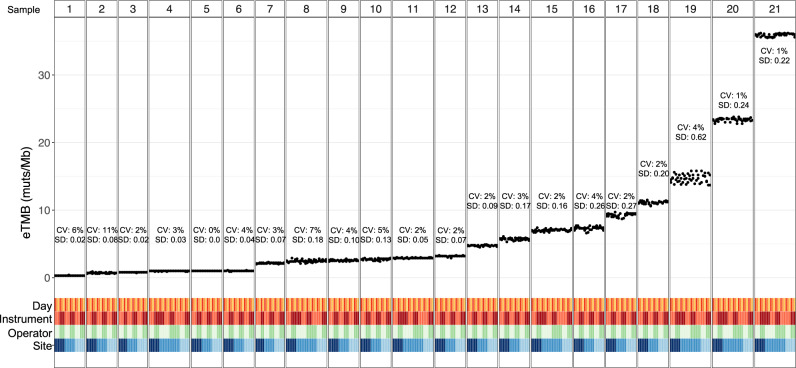
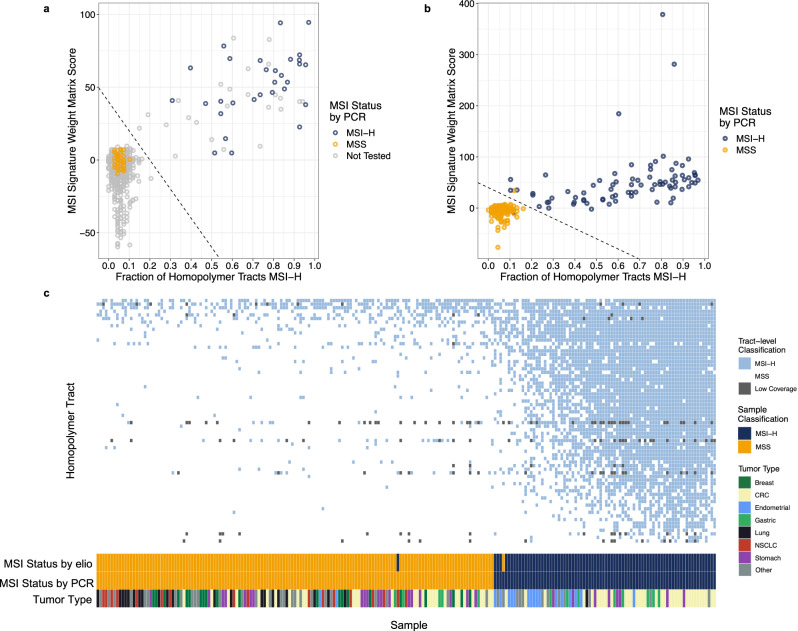
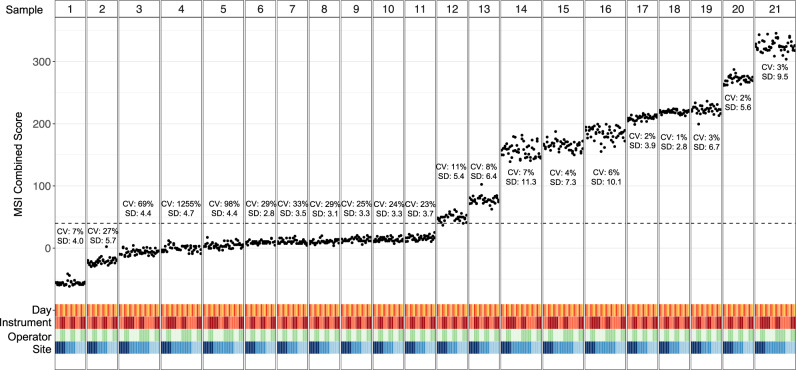
The lack of validated, distributed comprehensive genomic profiling assays for patients with cancer inhibits access to precision oncology treatment. To address this, we describe elio tissue complete, which has been FDA-cleared for examination of 505 cancer-related genes. Independent analyses of clinically and biologically relevant sequence changes across 170 clinical tumor samples using MSK-IMPACT, FoundationOne, and PCR-based methods reveals a positive percent agreement of >97%. We observe high concordance with whole-exome sequencing for evaluation of tumor mutational burden for 307 solid tumors (Pearson r = 0.95) and comparison of the elio tissue complete microsatellite instability detection approach with an independent PCR assay for 223 samples displays a positive percent agreement of 99%. Finally, evaluation of amplifications and translocations against DNA- and RNA-based approaches exhibits >98% negative percent agreement and positive percent agreement of 86% and 82%, respectively. These methods provide an approach for pan-solid tumor comprehensive genomic profiling with high analytical performance.
© 2022. The Author(s).
Conflict of interest statement
J.R.W. is the founder and owner of Resphera Biosciences LLC and serves as a consultant to Personal Genome Diagnostics. K.M.R.G., K.C.V., D.R., C.G., A.G., P.M.M., J.D., S.V.A., and M.S. are employees of Personal Genome Diagnostics. J.L.P. is an employee and stockholder of PathGroup. L.D. is a member of the board of directors of Jounce Therapeutics and Epitope. He is a compensated consultant to PetDx, Innovatus CP, Se’er, Delfi, Kinnate and Neophore. He is an inventor of multiple licensed patents (to Qiagen, Exact Biosciences, and LabCorp) related to technology for circulating tumor DNA analyses and mismatch repair deficiency for diagnosis and therapy. Some of these licenses and relationships are associated with equity or royalty payments to the inventors. He holds equity in Epitope, Jounce Therapeutics, PetDx, Se’er, Delfi, Kinnate and Neophore. He divested his equity in Personal Genome Diagnostics to LabCorp in February 2022 and divested his equity in Thrive Earlier Detection to Exact Biosciences in January 2021. His spouse holds equity in Amgen. The terms of all these arrangements are being managed by Memorial Sloan Kettering in accordance with their conflict-of-interest policy. V.E.V. is a founder of Delfi Diagnostics, serves on the Board of Directors and as a consultant for this organization, and owns Delfi Diagnostics stock, which is subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics. V.E.V. divested his equity in Personal Genome Diagnostics to LabCorp in February 2022. V.E.V. is an inventor on patent applications submitted by Johns Hopkins University related to cancer genomic analyses and cell-free DNA for cancer detection that have been licensed to one or more entities, including Delfi Diagnostics, LabCorp, Qiagen, Sysmex, Agios, Genzyme, Esoterix, Ventana and ManaT Bio. Under the terms of these license agreements, the University and inventors are entitled to fees and royalty distributions. V.E.V. is an advisor to Danaher, Takeda Pharmaceuticals, and Viron Therapeutics. These arrangements have been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies. The remaining authors declare no competing interests.
Figures
References
-
- Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. - DOI - PubMed
-
- Wagle N, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
