

Modeling of protein conformational changes with Rosetta guided by limited experimental data
- PMID: 35597243
- PMCID: PMC9357069
- DOI: 10.1016/j.str.2022.04.013
Modeling of protein conformational changes with Rosetta guided by limited experimental data
Abstract
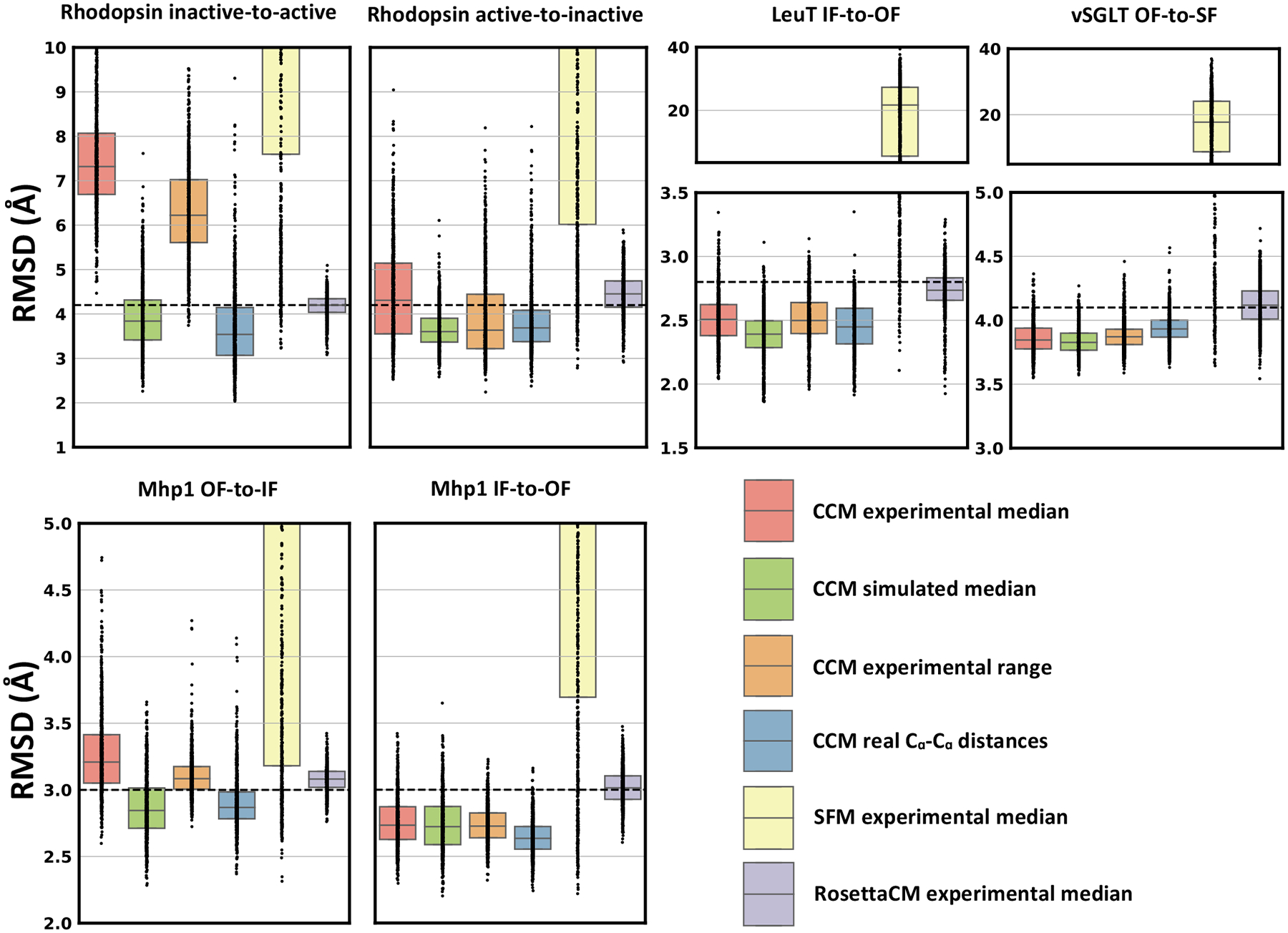
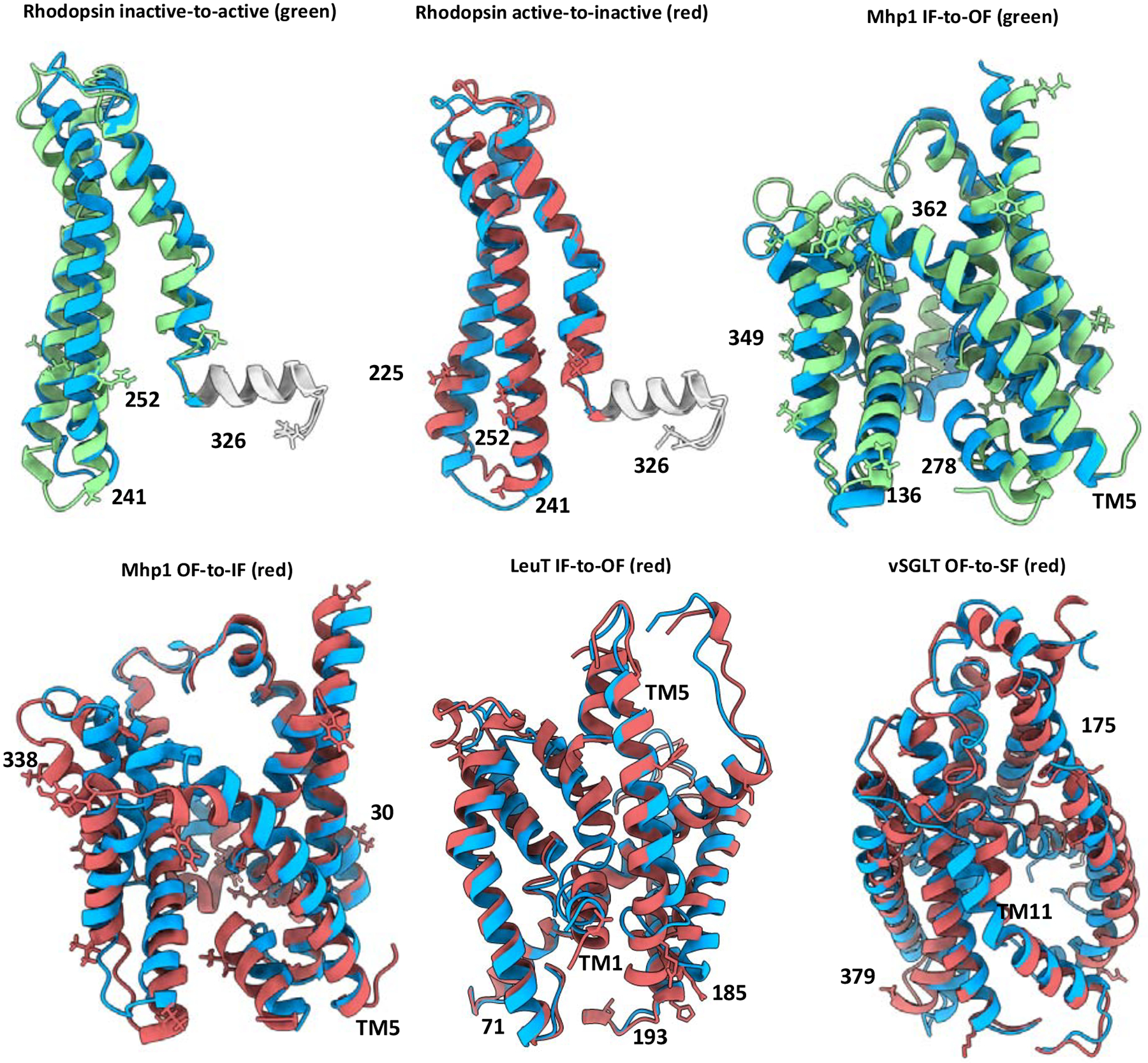
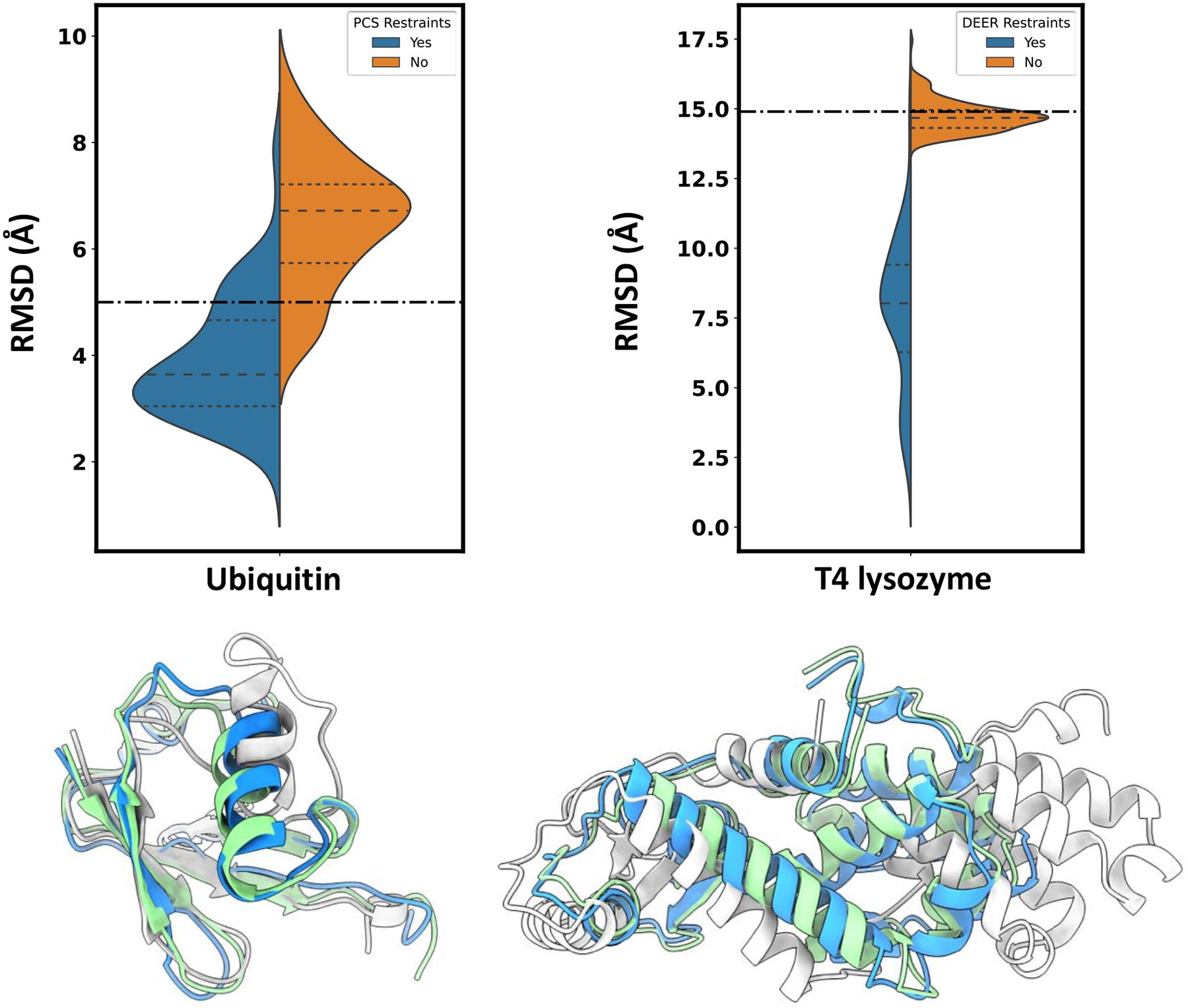
Conformational changes are an essential component of functional cycles of many proteins, but their characterization often requires an integrative structural biology approach. Here, we introduce and benchmark ConfChangeMover (CCM), a new method built into the widely used macromolecular modeling suite Rosetta that is tailored to model conformational changes in proteins using sparse experimental data. CCM can rotate and translate secondary structural elements and modify their backbone dihedral angles in regions of interest. We benchmarked CCM on soluble and membrane proteins with simulated Cα-Cα distance restraints and sparse experimental double electron-electron resonance (DEER) restraints, respectively. In both benchmarks, CCM outperformed state-of-the-art Rosetta methods, showing that it can model a diverse array of conformational changes. In addition, the Rosetta framework allows a wide variety of experimental data to be integrated with CCM, thus extending its capability beyond DEER restraints. This method will contribute to the biophysical characterization of protein dynamics.
Keywords: EPR spectroscopy; Rosetta; conformational changes; integrative structural biology; molecular modeling; protein dynamics; protein structure refinement.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
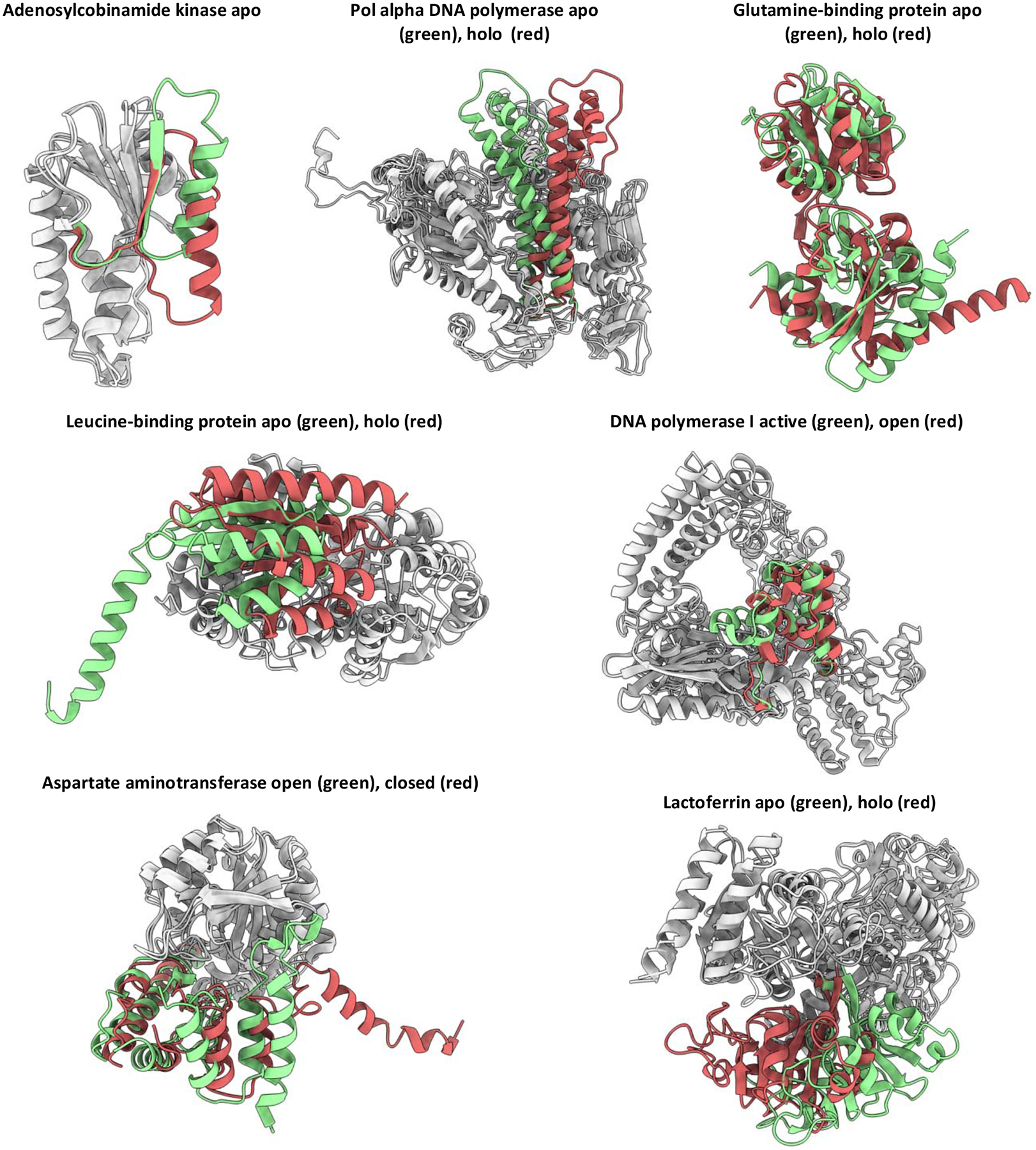
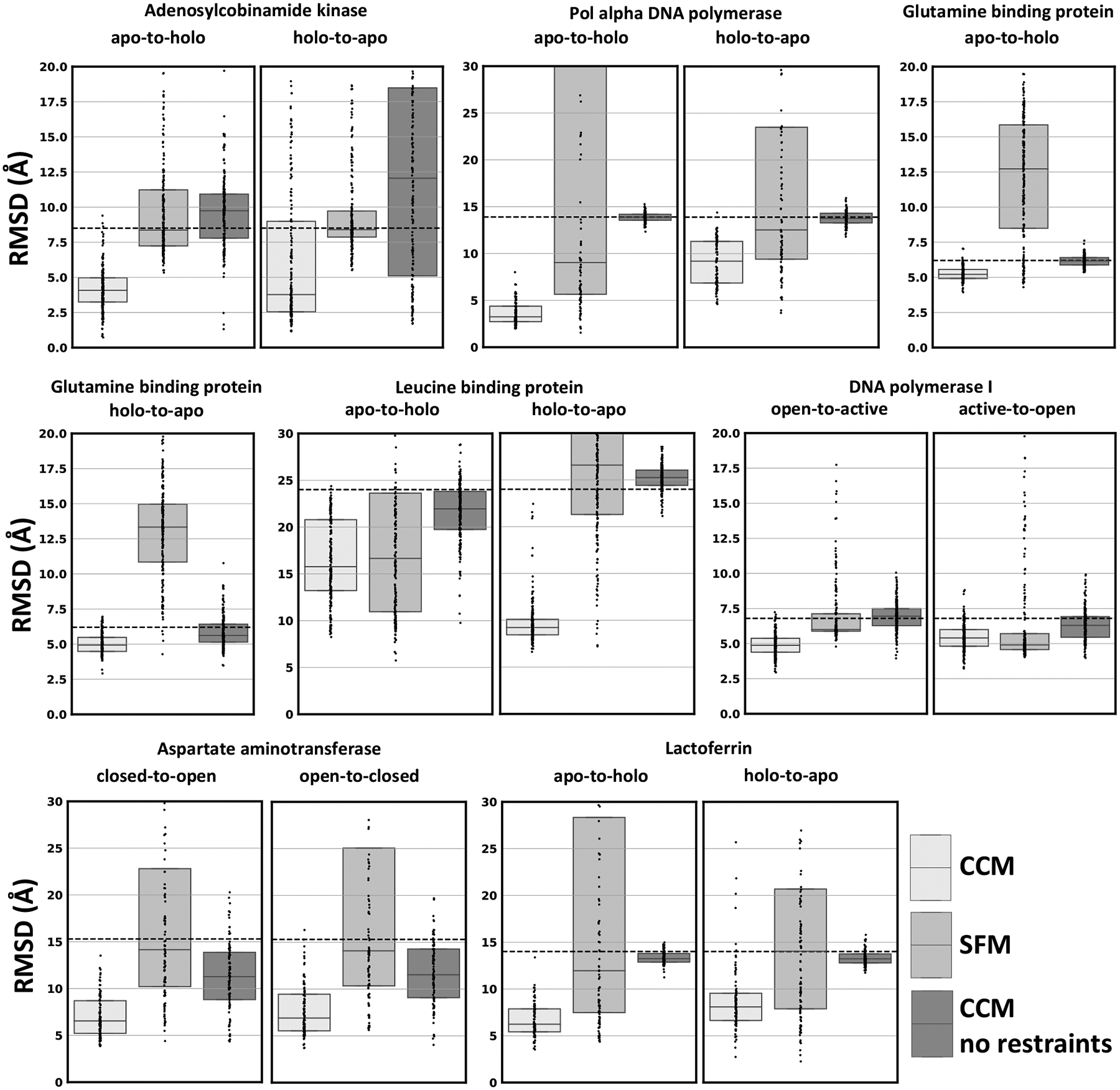
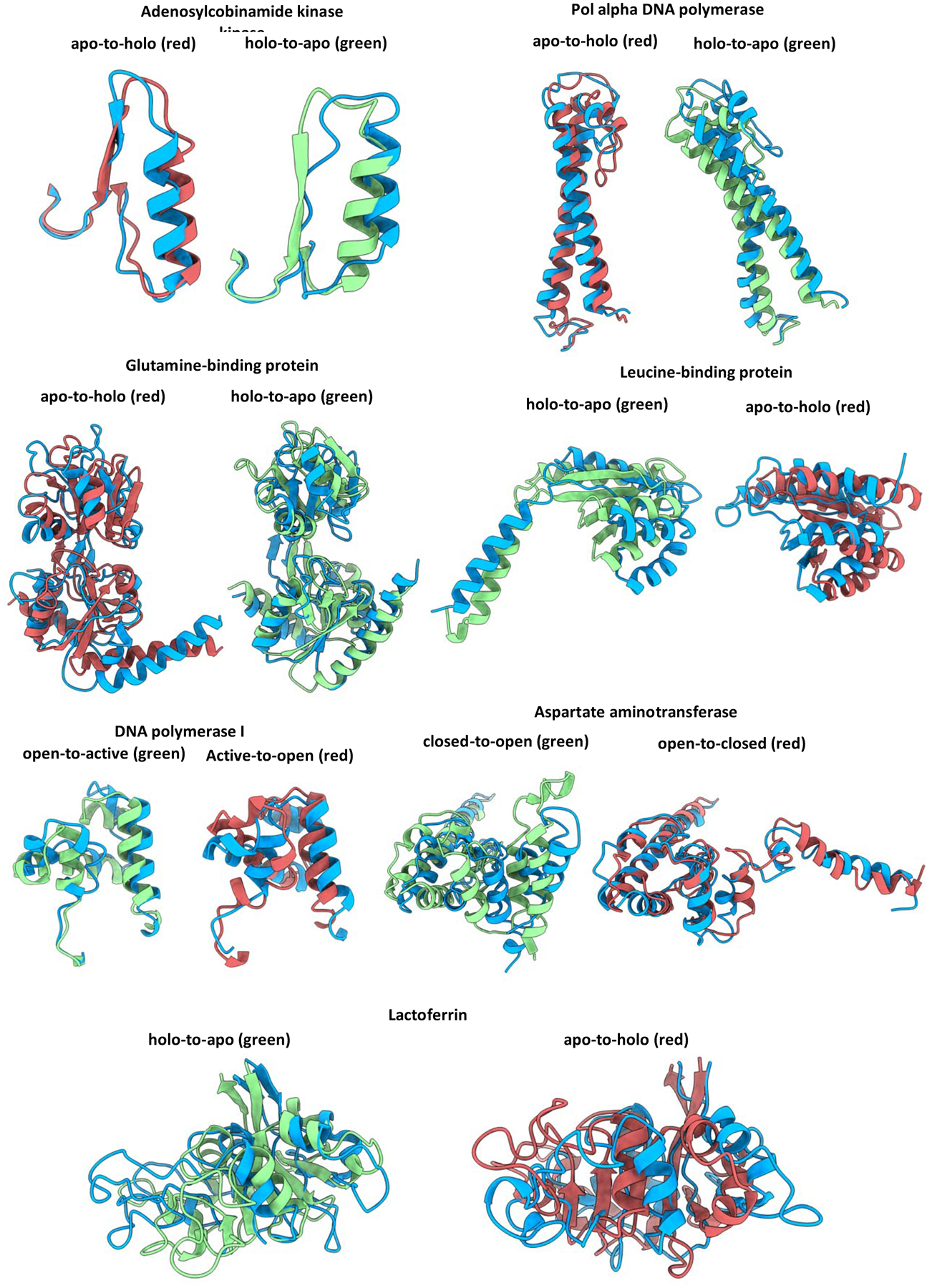
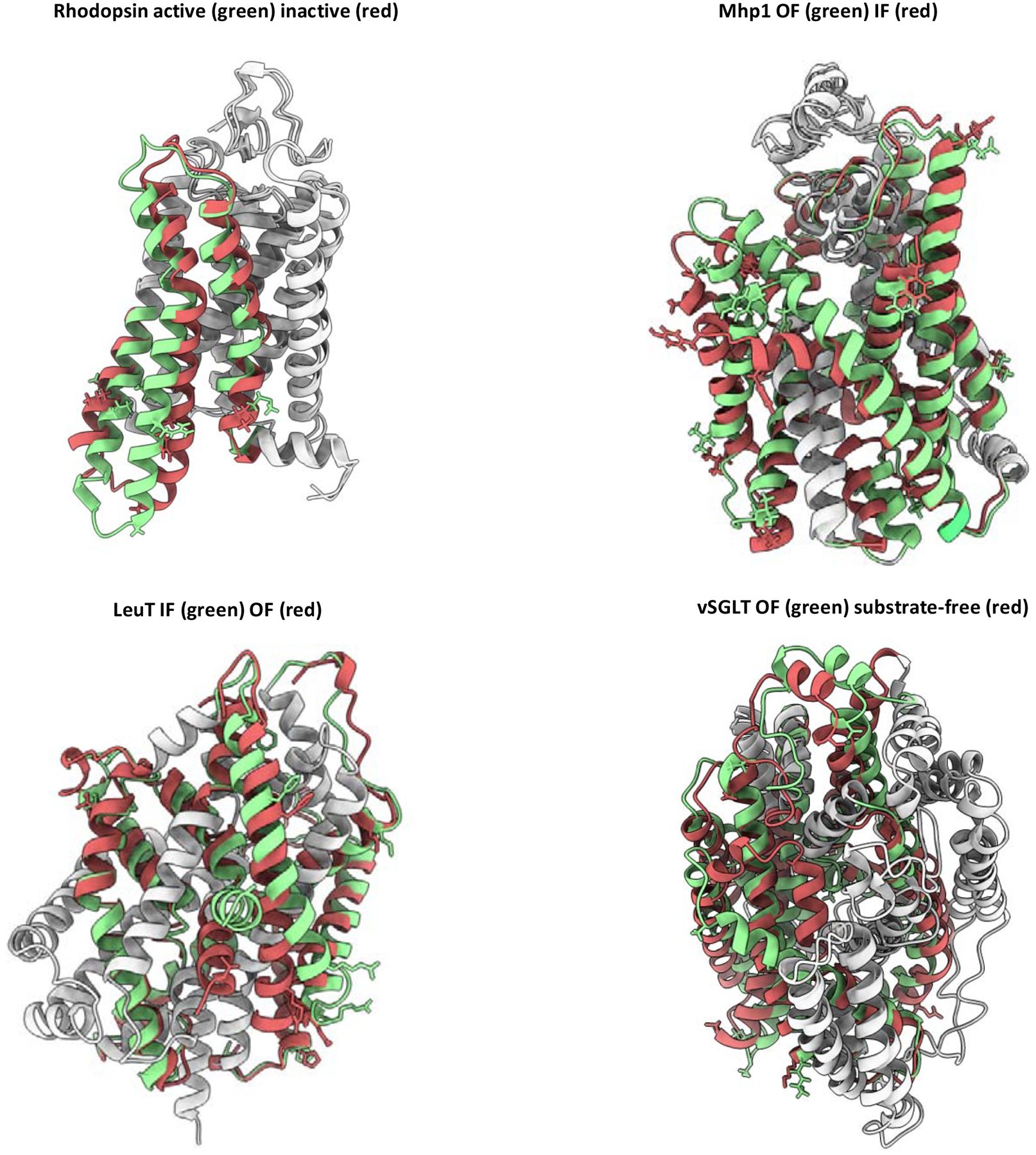
References
-
- del Alamo D, Fischer AW, Moretti R, Alexander NS, Mendenhall J, Hyman NJ, and Meiler J (2021b). Efficient Sampling of Protein Loop Regions Using Conformational Hashing Complemented with Random Coordinate Descent. J. Chem. Theory Comput 17, 560–570. - PubMed
-
- AlQuraishi M (2021). Machine learning in protein structure prediction. Curr. Opin. Chem. Biol 65, 1–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
