

Cancer cell cycle dystopia: heterogeneity, plasticity, and therapy
- PMID: 35599231
- PMCID: PMC9388619
- DOI: 10.1016/j.trecan.2022.04.006
Cancer cell cycle dystopia: heterogeneity, plasticity, and therapy
Abstract
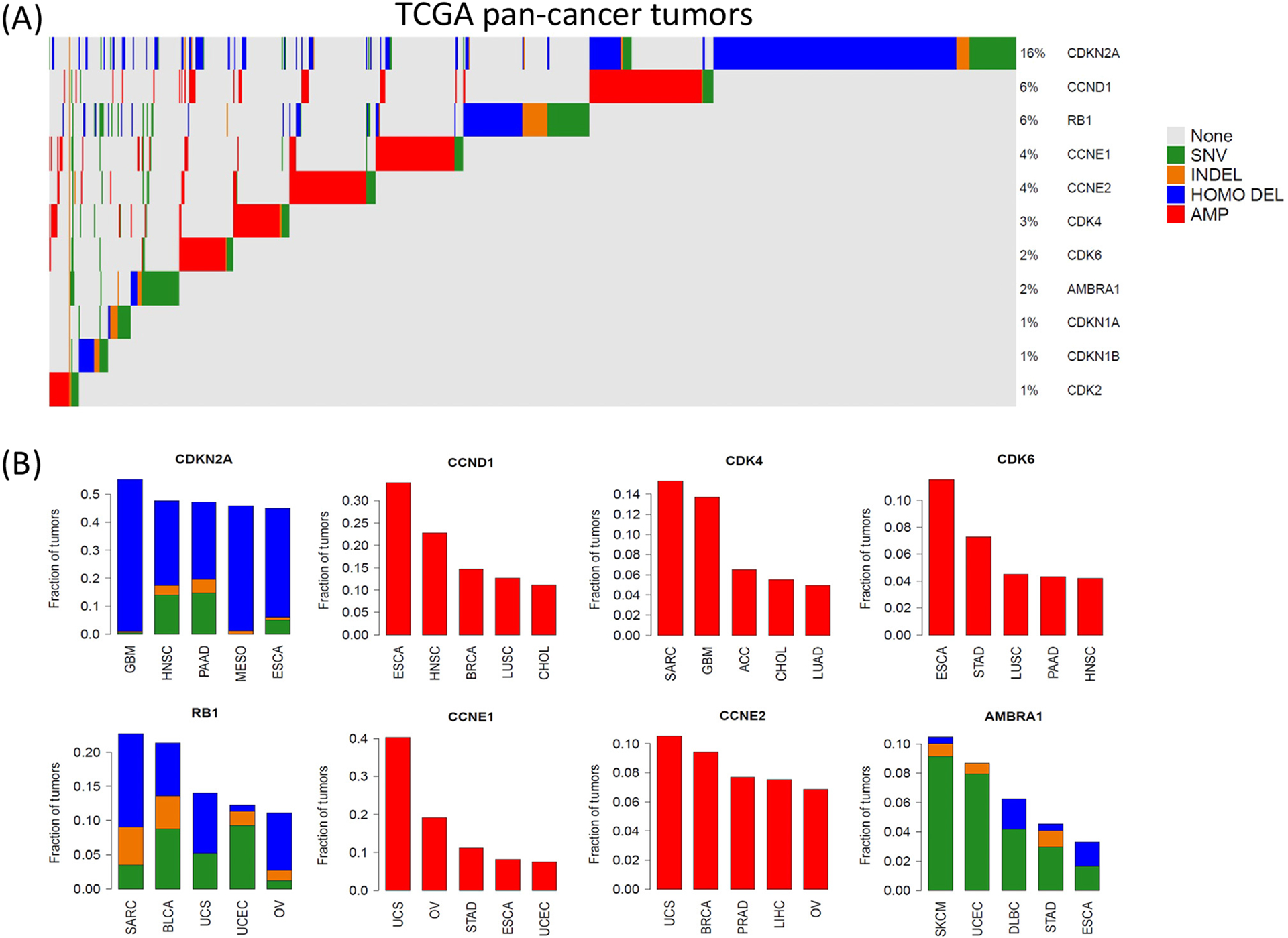
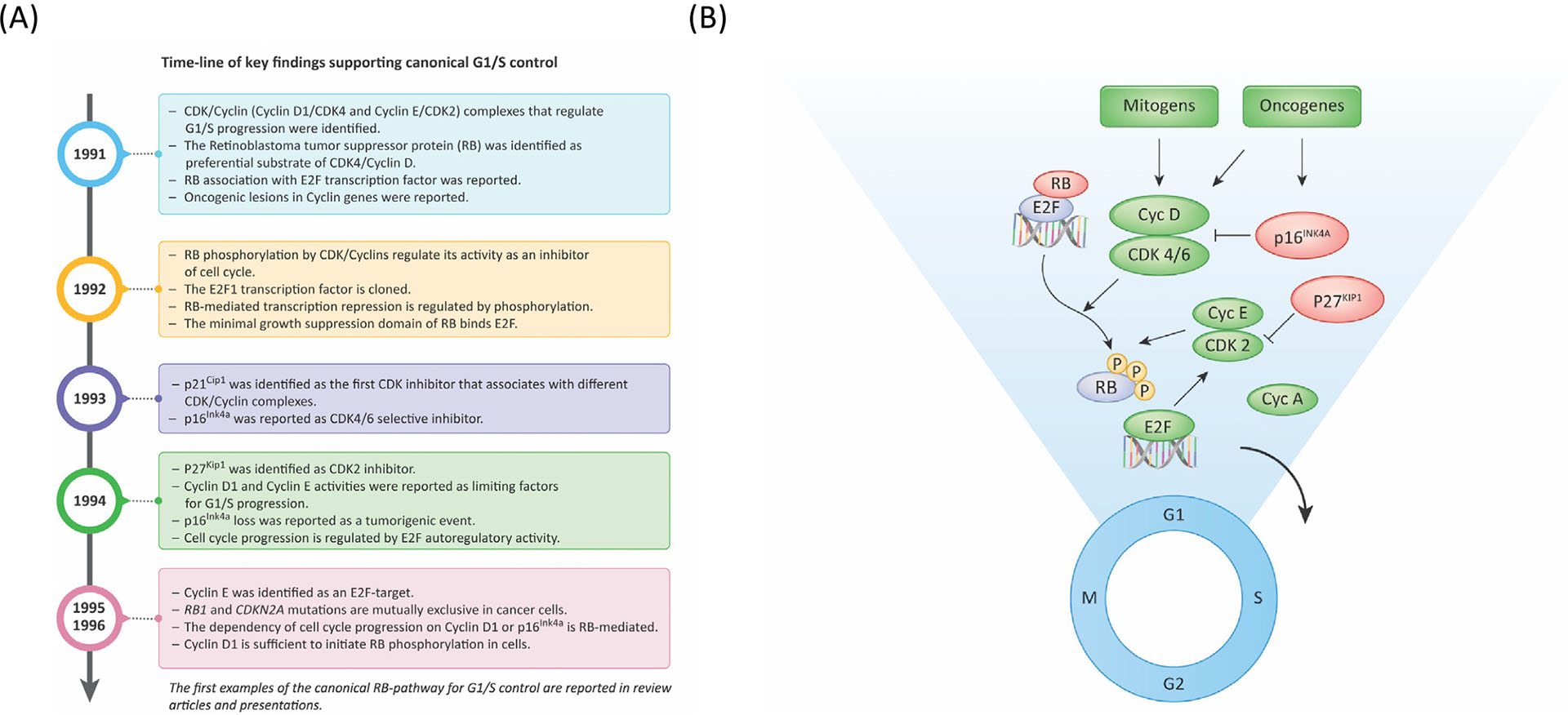
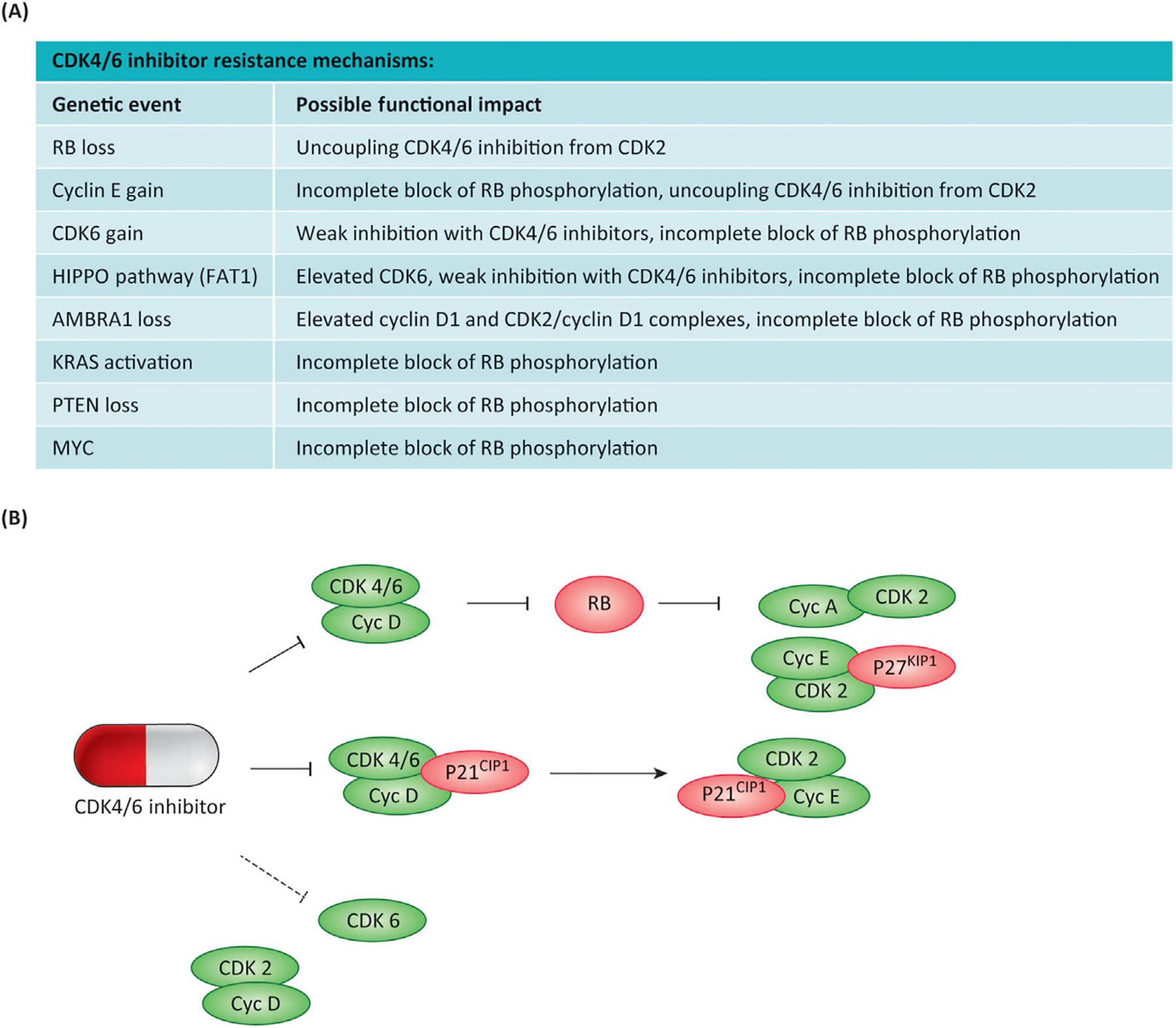
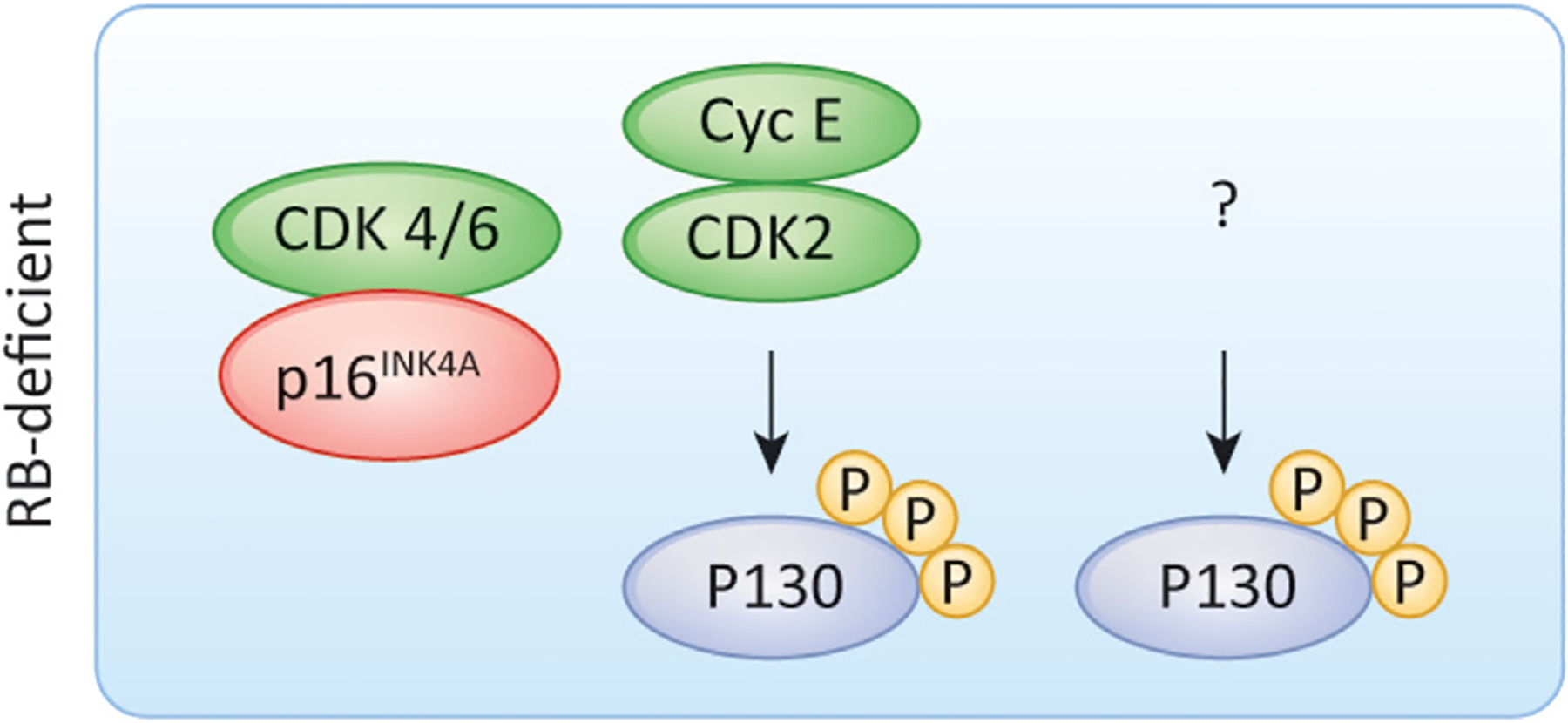
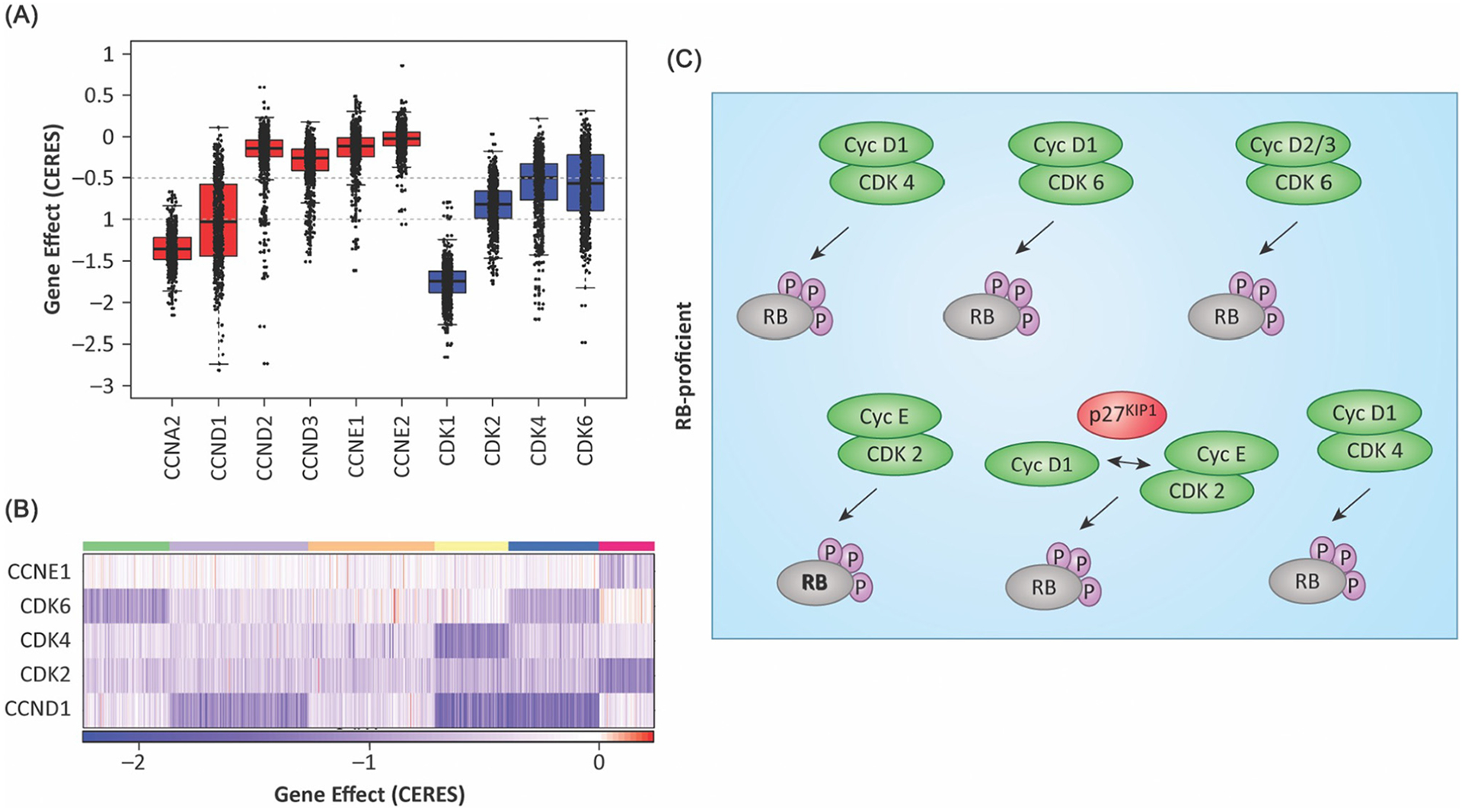
The mammalian cell cycle has been extensively studied regarding cancer etiology, progression, and therapeutic intervention. The canonical cell cycle framework is supported by a plethora of data pointing to a relatively simple linear pathway in which mitogenic signals are integrated in a stepwise fashion to allow progression through G1/S with coordinate actions of cyclin-dependent kinases (CDK)4/6 and CDK2 on the RB tumor suppressor. Recent work on adaptive mechanisms and intrinsic heterogeneous dependencies indicates that G1/S control of the cell cycle is a variable signaling pathway rather than an invariant engine that drives cell division. These alterations can limit the effectiveness of pharmaceutical agents but provide new avenues for therapeutic interventions. These findings support a dystopian view of the cell cycle in cancer where the canonical utopian cell cycle is often not observed. However, recognizing the extent of cell cycle heterogeneity likely creates new opportunities for precision therapeutic approaches specifically targeting these states.
Keywords: CDK4; CDK6; CHK1; RB; abemaciclib; ambra1; aurora kinase; cyclin D1; cyclin E; p16(INK4A); p27(KIP1); palbociclib.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
