

Identification of a Potential mRNA-based Vaccine Candidate against the SARS-CoV-2 Spike Glycoprotein: A Reverse Vaccinology Approach
- PMID: 35601809
- PMCID: PMC9111088
- DOI: 10.1002/slct.202103903
Identification of a Potential mRNA-based Vaccine Candidate against the SARS-CoV-2 Spike Glycoprotein: A Reverse Vaccinology Approach
Abstract
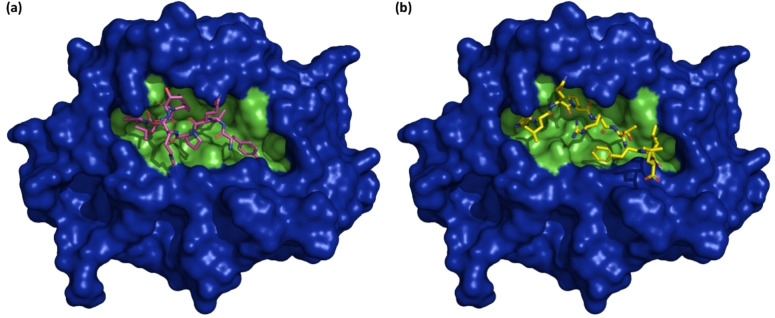
The emergence of the novel coronavirus (SARS-CoV-2) in December 2019 has generated a devastating global consequence which makes the development of a rapidly deployable, effective and safe vaccine candidate an imminent global health priority. The design of most vaccine candidates has been directed at the induction of antibody responses against the trimeric spike glycoprotein of SARS-CoV-2, a class I fusion protein that aids ACE2 (angiotensin-converting enzyme 2) receptor binding. A variety of formulations and vaccinology approaches are being pursued for targeting the spike glycoprotein, including simian and human replication-defective adenoviral vaccines, subunit protein vaccines, nucleic acid vaccines and whole-inactivated SARS-CoV-2. Here, we directed a reverse vaccinology approach towards the design of a nucleic acid (mRNA-based) vaccine candidate. The "YLQPRTFLL" peptide sequence (position 269-277) which was predicted to be a B cell epitope and likewise a strong binder of the HLA*A-0201 was selected for the design of the vaccine candidate, having satisfied series of antigenicity assessments. Through the codon optimization protocol, the nucleotide sequence for the vaccine candidate design was generated and targeted at the human toll-like receptor 7 (TLR7). Bioinformatics analyses showed that the sequence "UACCUGCAGCCGCGUACCUUCCUGCUG" exhibited a strong affinity and likewise was bound to a stable cavity in the TLR7 pocket. This study is therefore expected to contribute to the research efforts directed at securing definitive preventive measures against the SARS-CoV-2 infection.
Keywords: Antigens; Reverse vaccinology; SAR-CoV-2; Spike glycoprotein; Vaccine; Viruses.
© 2022 Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures















References
-
- Drosten C., Günther S., Preiser W., Van Der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R. A., N. Engl. J. Med. 2003, 348 (20), 1967. - PubMed
-
- Zaki A. M., Van Boheemen S., Bestebroer T. M., Osterhaus A. D., Fouchier R. A., N. Engl. J. Med. 2012, 367 (19), 1814. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous