

STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity
- PMID: 35606429
- PMCID: PMC9156412
- DOI: 10.1038/s41563-022-01251-z
STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity
Abstract
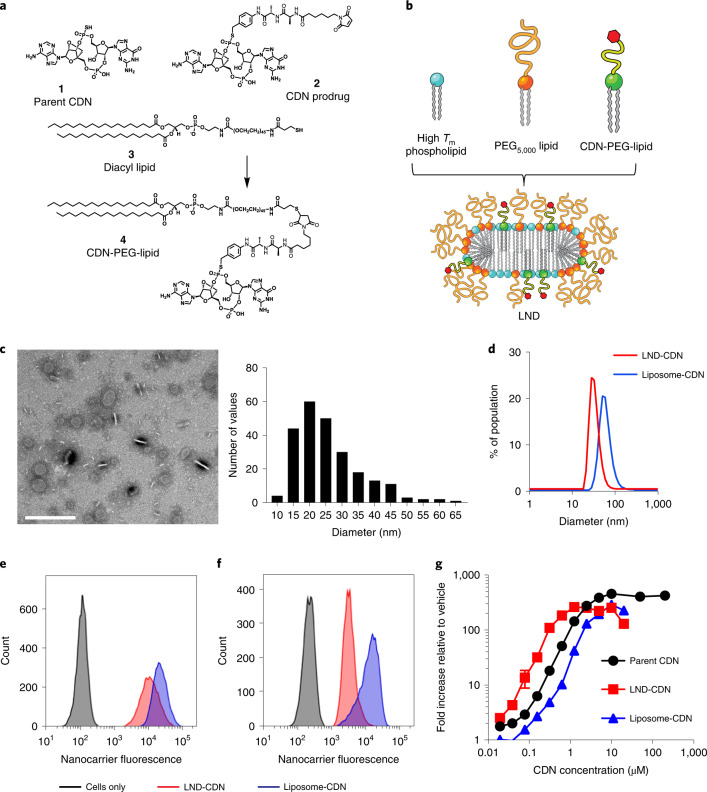
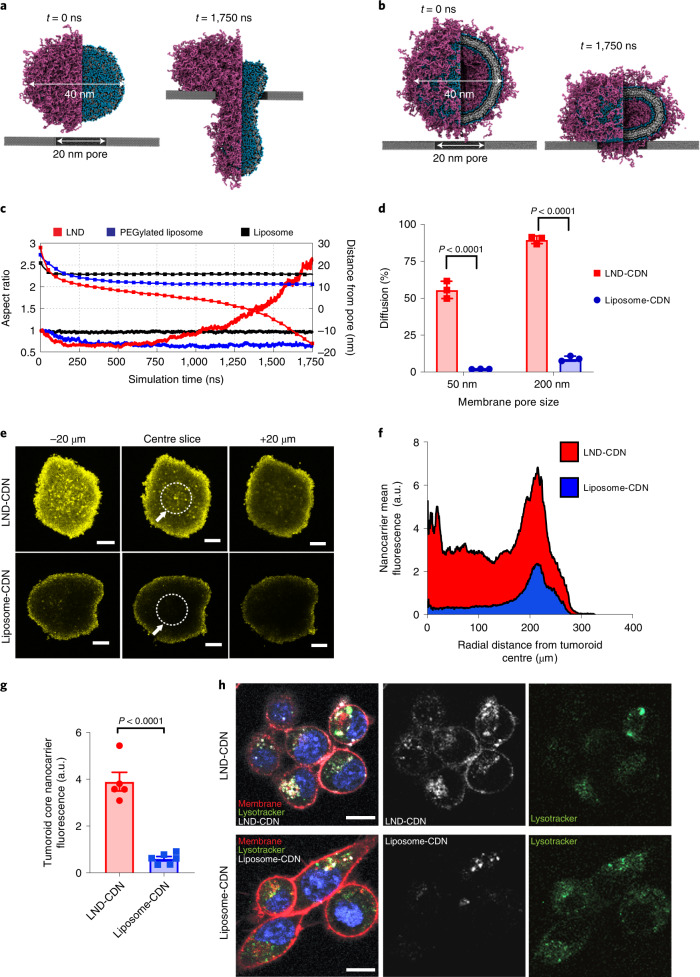
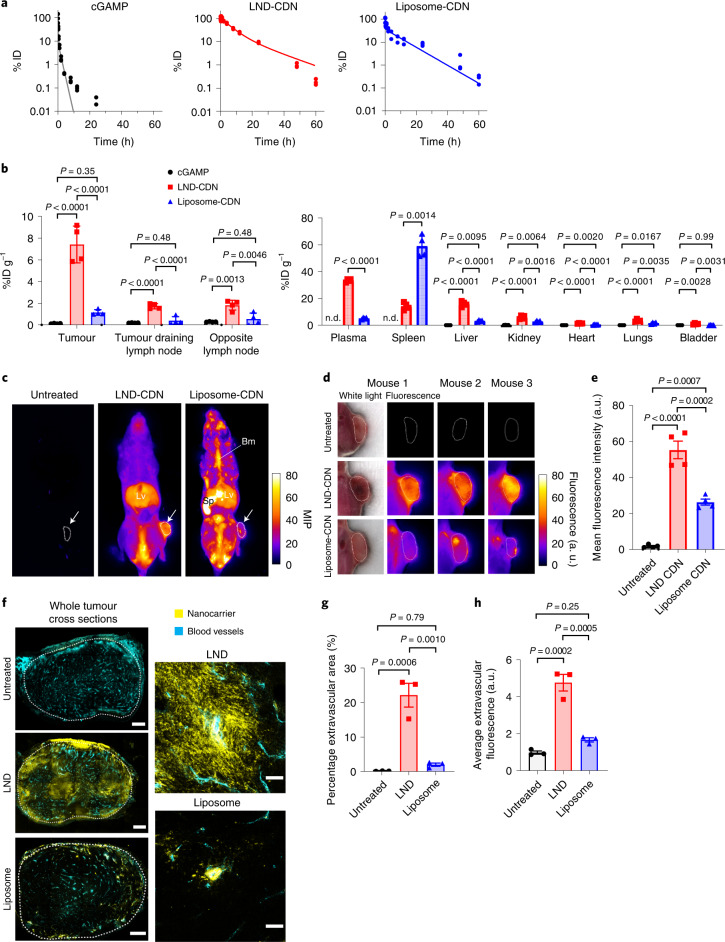
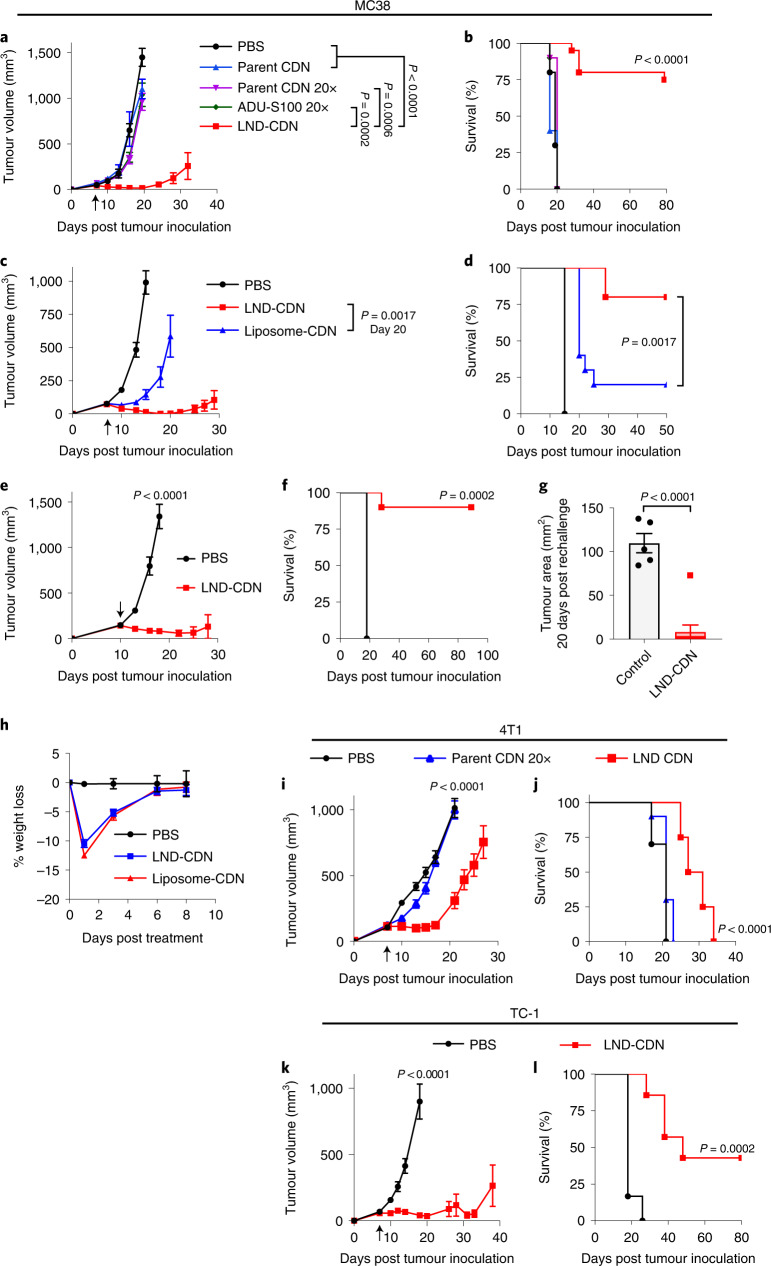
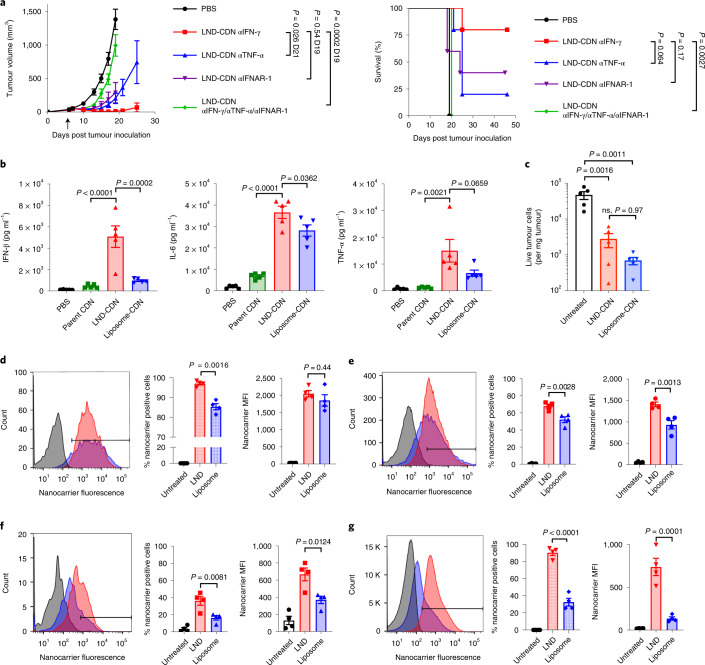
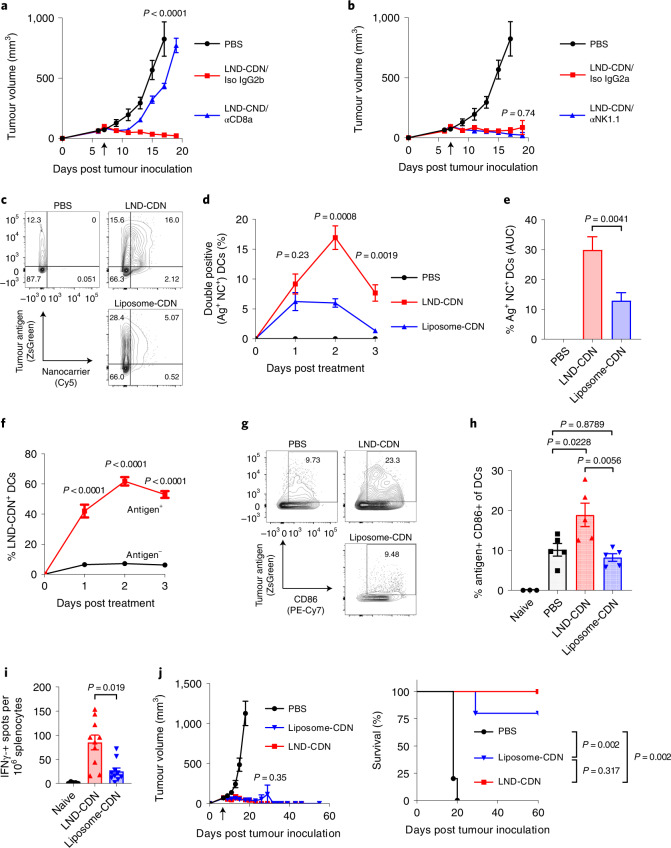
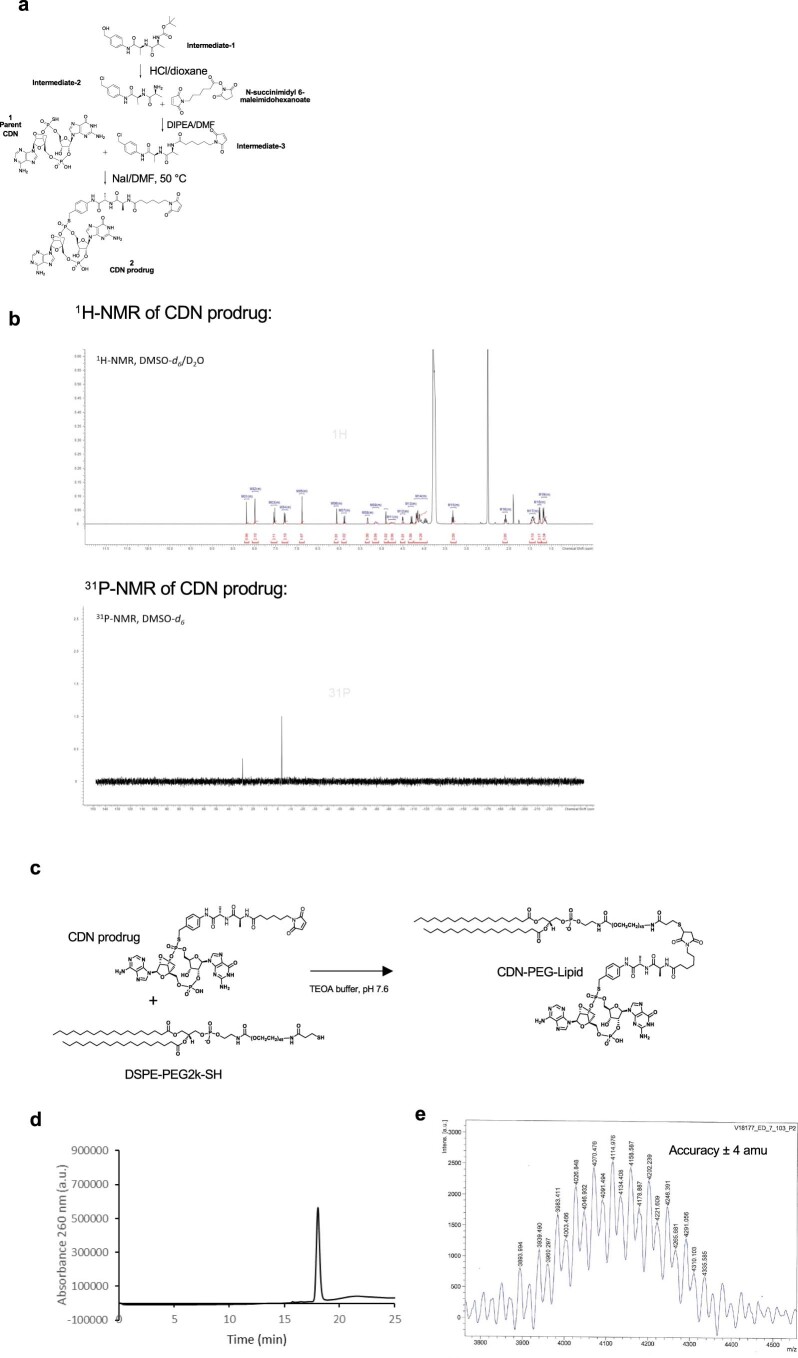
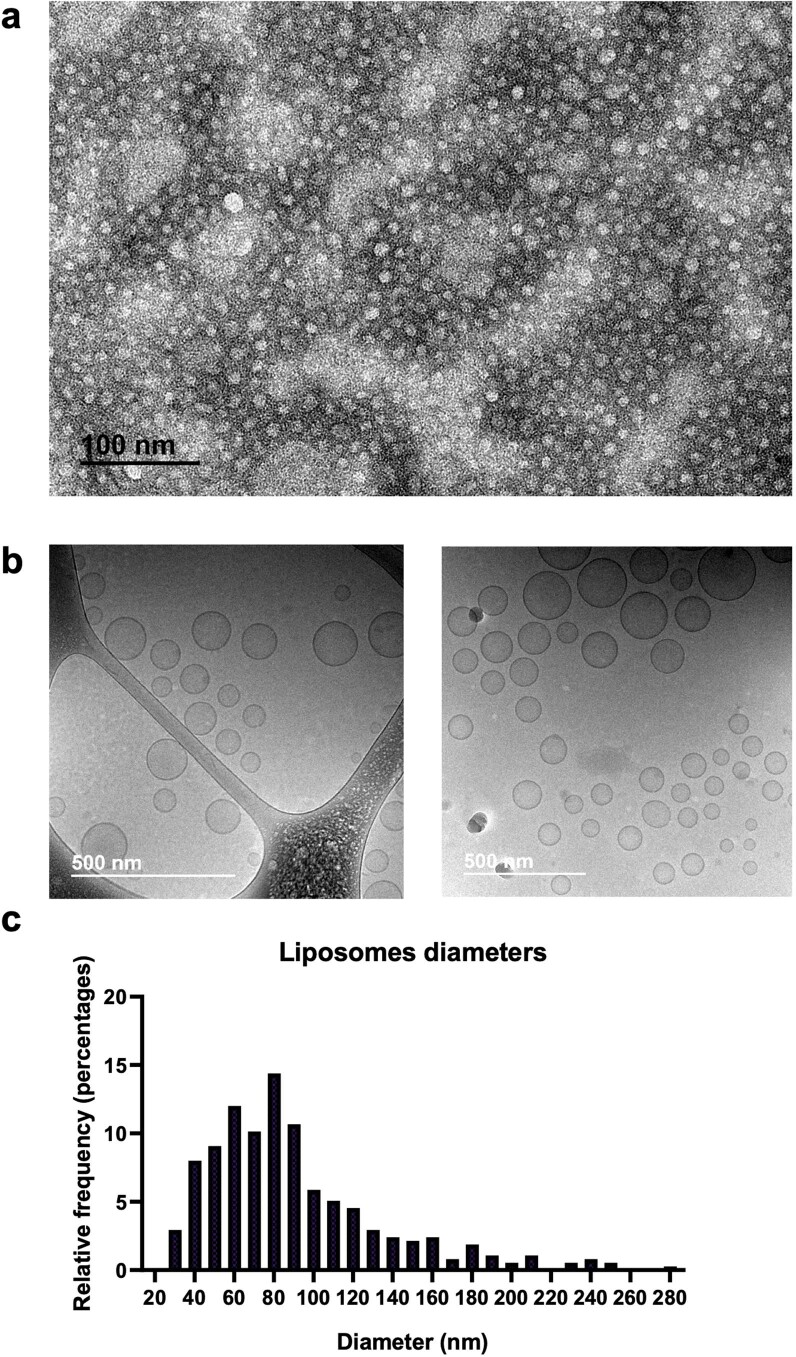
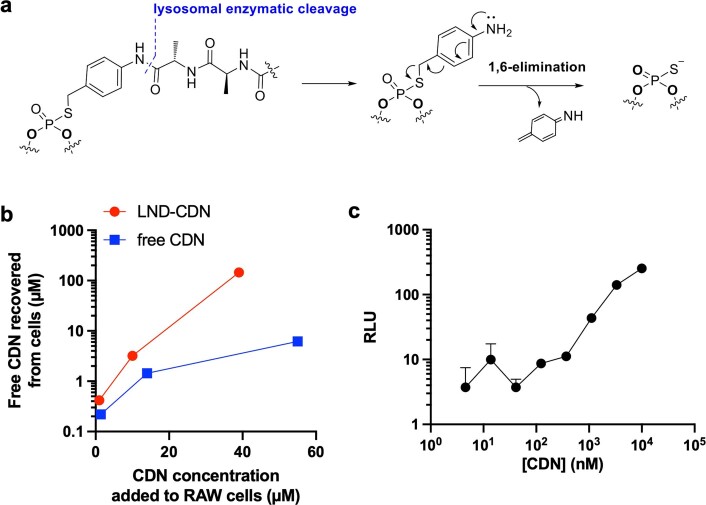
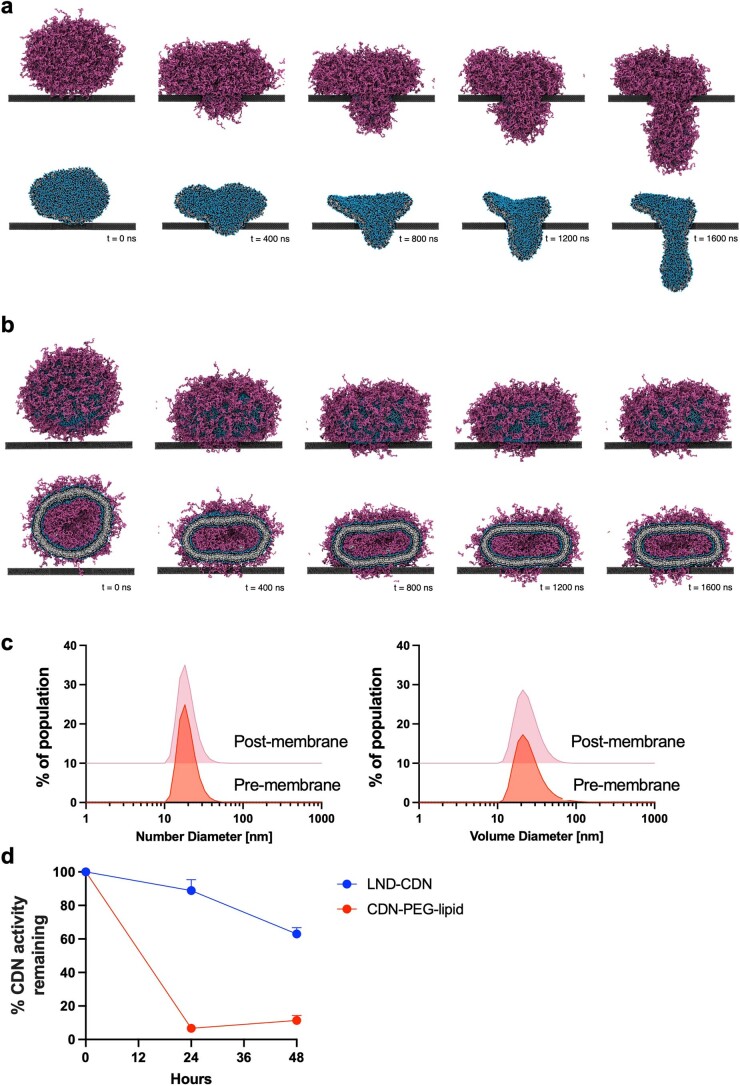
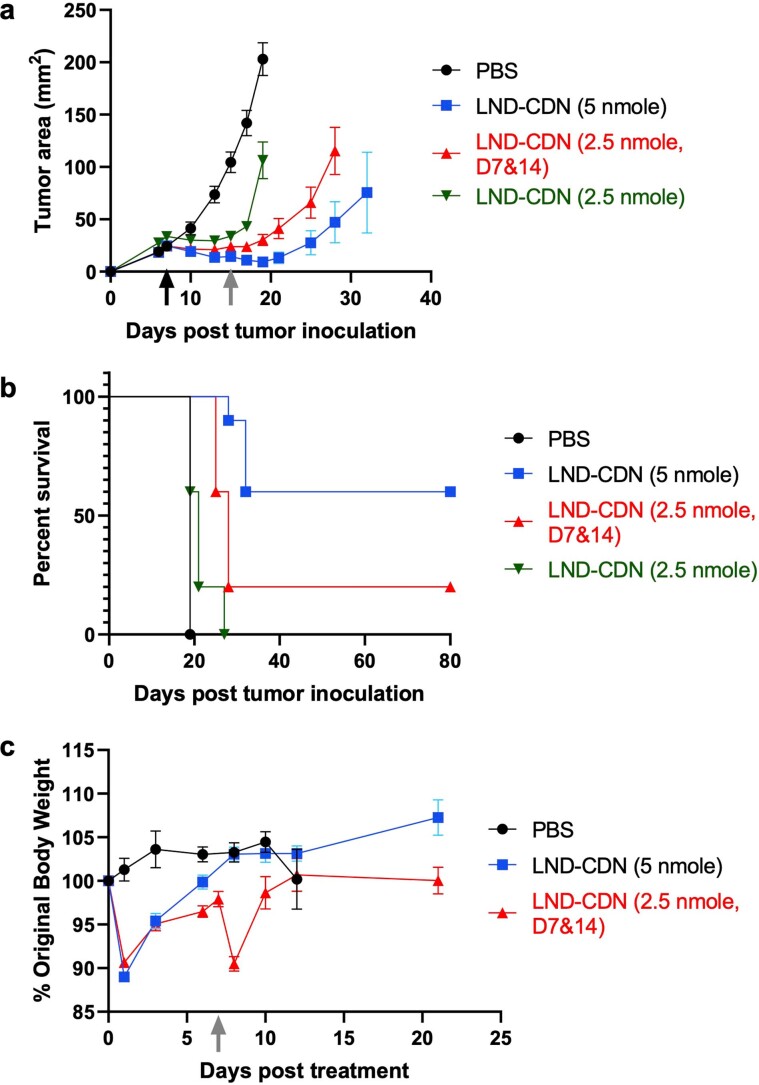
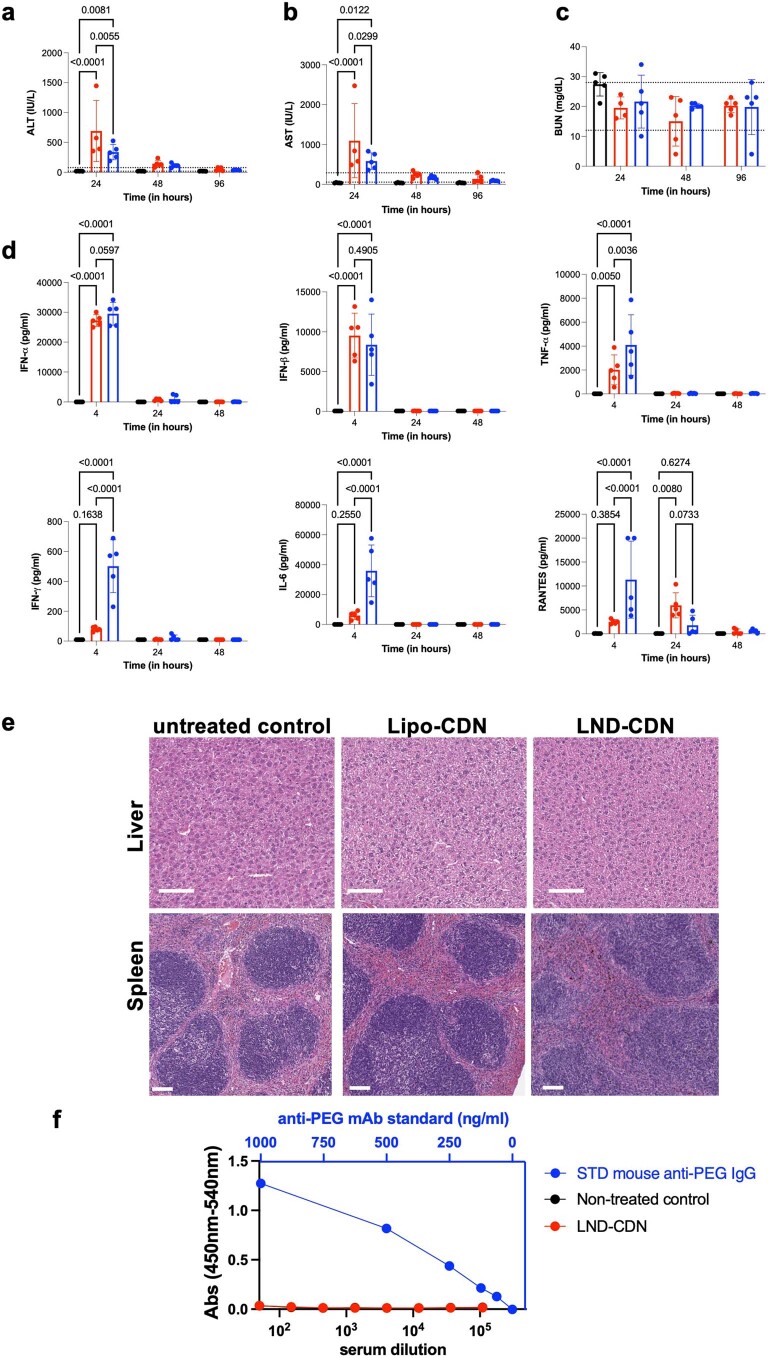
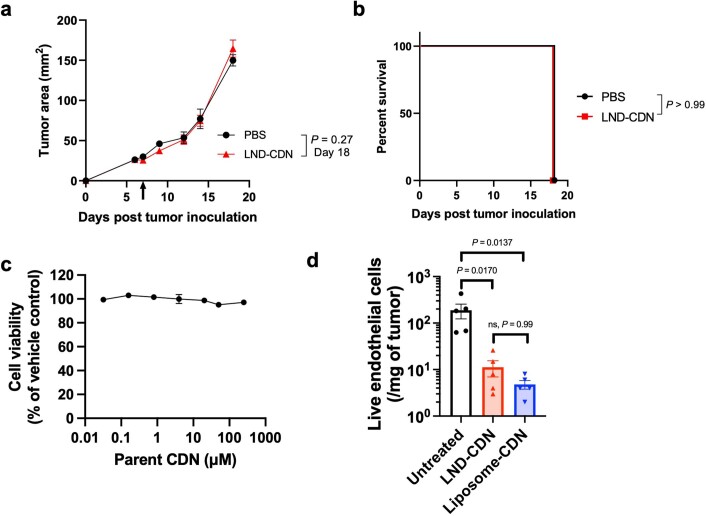
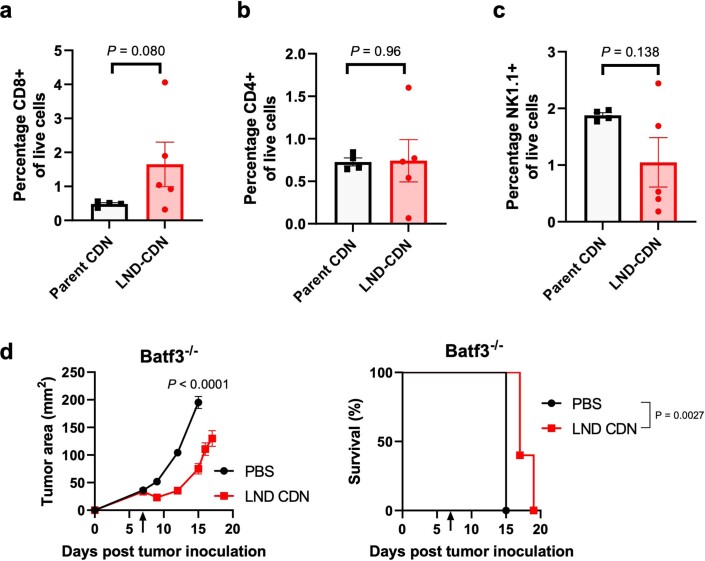
Activation of the innate immune STimulator of INterferon Genes (STING) pathway potentiates antitumour immunity, but systemic delivery of STING agonists to tumours is challenging. We conjugated STING-activating cyclic dinucleotides (CDNs) to PEGylated lipids (CDN-PEG-lipids; PEG, polyethylene glycol) via a cleavable linker and incorporated them into lipid nanodiscs (LNDs), which are discoid nanoparticles formed by self-assembly. Compared to state-of-the-art liposomes, intravenously administered LNDs carrying CDN-PEG-lipid (LND-CDNs) exhibited more efficient penetration of tumours, exposing the majority of tumour cells to STING agonist. A single dose of LND-CDNs induced rejection of established tumours, coincident with immune memory against tumour rechallenge. Although CDNs were not directly tumoricidal, LND-CDN uptake by cancer cells correlated with robust T-cell activation by promoting CDN and tumour antigen co-localization in dendritic cells. LNDs thus appear promising as a vehicle for robust delivery of compounds throughout solid tumours, which can be exploited for enhanced immunotherapy.
© 2022. The Author(s).
Conflict of interest statement
D.J.I., E.L.D. and M.O. are inventors on a patent related to the LND-CDN technology. The remaining authors declare no competing interests.
Figures
Comment in
-
Lipid nanodiscs give cancer a STING.Nat Mater. 2022 Jun;21(6):616-617. doi: 10.1038/s41563-022-01270-w. Nat Mater. 2022. PMID: 35606430 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
