

Shortcutting the diagnostic odyssey: the multidisciplinary Program for Undiagnosed Rare Diseases in adults (UD-PrOZA)
- PMID: 35606766
- PMCID: PMC9128245
- DOI: 10.1186/s13023-022-02365-y
Shortcutting the diagnostic odyssey: the multidisciplinary Program for Undiagnosed Rare Diseases in adults (UD-PrOZA)
Abstract
Background: In order to facilitate the diagnostic process for adult patients suffering from a rare disease, the Undiagnosed Disease Program (UD-PrOZA) was founded in 2015 at the Ghent University Hospital in Belgium. In this study we report the five-year results of our multidisciplinary approach in rare disease diagnostics.
Methods: Patients referred by a healthcare provider, in which an underlying rare disease is likely, qualify for a UD-PrOZA evaluation. UD-PrOZA uses a multidisciplinary clinical approach combined with state-of-the-art genomic technologies in close collaboration with research facilities to diagnose patients.
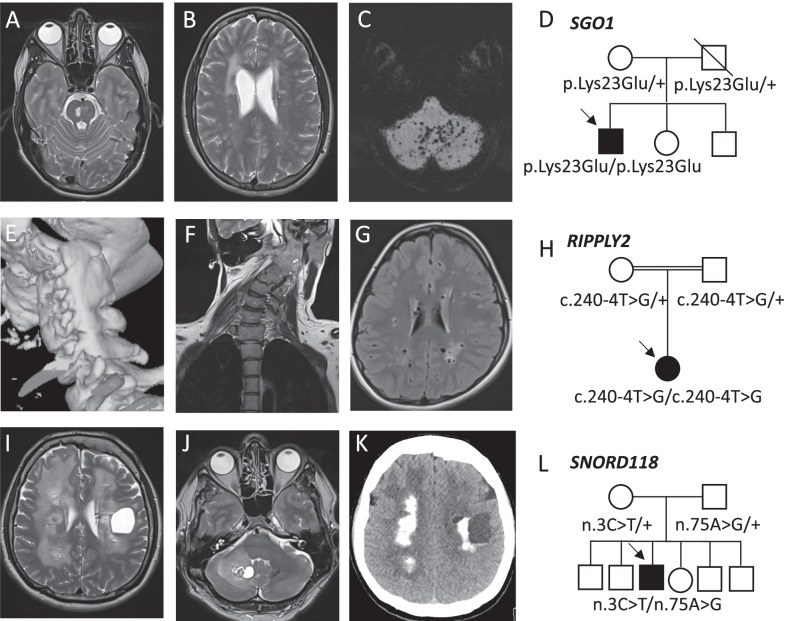
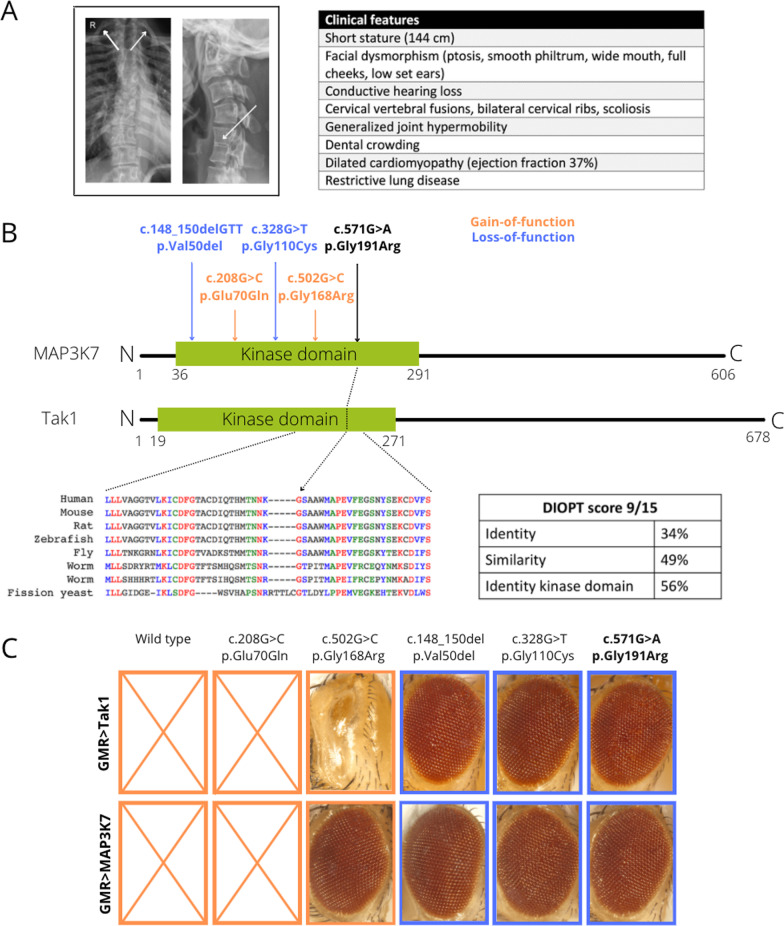
Results: Between 2015 and 2020, 692 patients (94% adults) were referred of which 329 (48%) were accepted for evaluation. In 18% (60 of 329) of the cases a definite diagnosis was made. 88% (53 of 60) of the established diagnoses had a genetic origin. 65% (39 of 60) of the genetic diagnoses were made through whole exome sequencing (WES). The mean time interval between symptom-onset and diagnosis was 19 years. Key observations included novel genotype-phenotype correlations, new variants in known disease genes and the identification of three new disease genes. In 13% (7 of 53), identifying the molecular cause was associated with therapeutic recommendations and in 88% (53 of 60), gene specific genetic counseling was made possible. Actionable secondary findings were reported in 7% (12 of 177) of the patients in which WES was performed.
Conclusion: UD-PrOZA offers an innovative interdisciplinary platform to diagnose rare diseases in adults with previously unexplained medical problems and to facilitate translational research.
Keywords: ACMSD; Diagnostic odyssey; Diagnostic yield; IRF2BPL; MAP3K7; PLAAT3; Rare diseases; SGO1; SNORD118; UD-PrOZA; Whole exome sequencing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Rath A, Salamon V, Peixoto S, Hivert V, Laville M, Segrestin B, et al. A systematic literature review of evidence-based clinical practice for rare diseases: what are the perceived and real barriers for improving the evidence and how can they be overcome? Trials. 2017;18(1):556. doi: 10.1186/s13063-017-2287-7. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
