

Comprehensive assessment of differential ChIP-seq tools guides optimal algorithm selection
- PMID: 35606795
- PMCID: PMC9128273
- DOI: 10.1186/s13059-022-02686-y
Comprehensive assessment of differential ChIP-seq tools guides optimal algorithm selection
Abstract
Background: The analysis of chromatin binding patterns of proteins in different biological states is a main application of chromatin immunoprecipitation followed by sequencing (ChIP-seq). A large number of algorithms and computational tools for quantitative comparison of ChIP-seq datasets exist, but their performance is strongly dependent on the parameters of the biological system under investigation. Thus, a systematic assessment of available computational tools for differential ChIP-seq analysis is required to guide the optimal selection of analysis tools based on the present biological scenario.
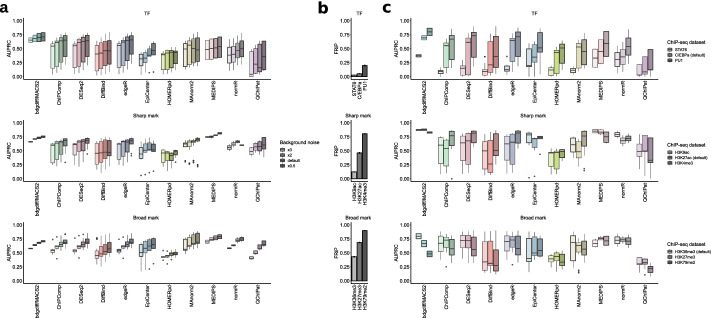
Results: We created standardized reference datasets by in silico simulation and sub-sampling of genuine ChIP-seq data to represent different biological scenarios and binding profiles. Using these data, we evaluated the performance of 33 computational tools and approaches for differential ChIP-seq analysis. Tool performance was strongly dependent on peak size and shape as well as on the scenario of biological regulation.
Conclusions: Our analysis provides unbiased guidelines for the optimized choice of software tools in differential ChIP-seq analysis.
Keywords: Benchmarking differential ChIP-seq tools; Bioinformatic analysis; Differential ChIP-seq; Guidelines for differential ChIP-seq.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures







References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
