

An automated multi-modal graph-based pipeline for mouse genetic discovery
- PMID: 35608290
- PMCID: PMC9992076
- DOI: 10.1093/bioinformatics/btac356
An automated multi-modal graph-based pipeline for mouse genetic discovery
Abstract
Motivation: Our ability to identify causative genetic factors for mouse genetic models of human diseases and biomedical traits has been limited by the difficulties associated with identifying true causative factors, which are often obscured by the many false positive genetic associations produced by a GWAS.
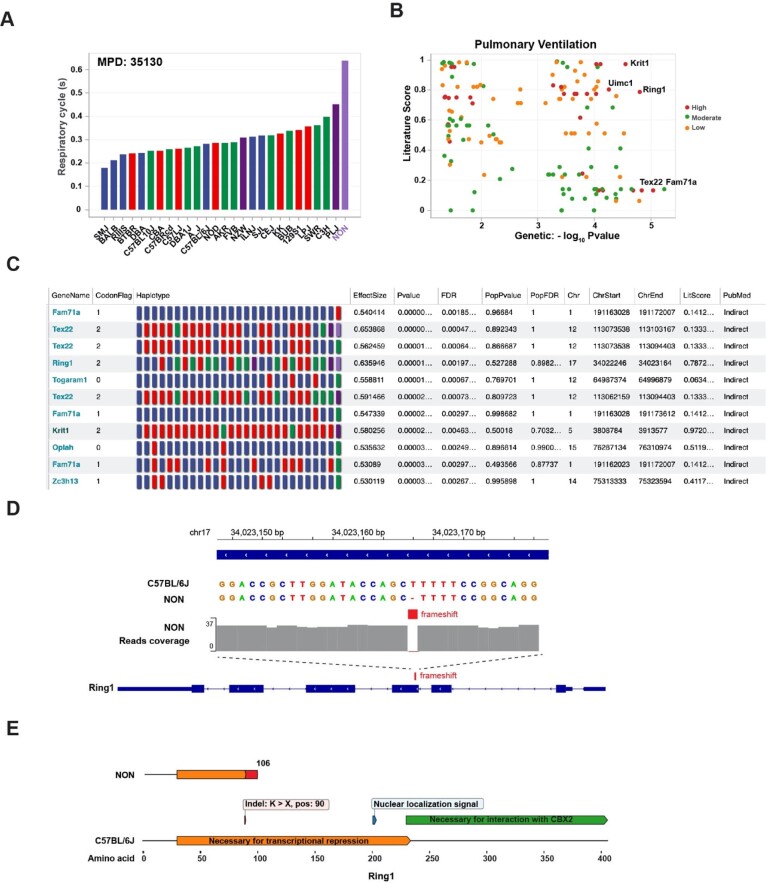
Results: To accelerate the pace of genetic discovery, we developed a graph neural network (GNN)-based automated pipeline (GNNHap) that could rapidly analyze mouse genetic model data and identify high probability causal genetic factors for analyzed traits. After assessing the strength of allelic associations with the strain response pattern; this pipeline analyzes 29M published papers to assess candidate gene-phenotype relationships; and incorporates the information obtained from a protein-protein interaction network and protein sequence features into the analysis. The GNN model produces markedly improved results relative to that of a simple linear neural network. We demonstrate that GNNHap can identify novel causative genetic factors for murine models of diabetes/obesity and for cataract formation, which were validated by the phenotypes appearing in previously analyzed gene knockout mice. The diabetes/obesity results indicate how characterization of the underlying genetic architecture enables new therapies to be discovered and tested by applying 'precision medicine' principles to murine models.
Availability and implementation: The GNNHap source code is freely available at https://github.com/zqfang/gnnhap, and the new version of the HBCGM program is available at https://github.com/zqfang/haplomap.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures




References
-
- Agrawal S. et al. (2003) Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J. Immunol., 171, 4984–4989. - PubMed
-
- Arslan A. et al. (2020) High throughput computational mouse genetic analysis. BioRxiv. https://www.biorxiv.org/content/10.1101/2020.09.01.278465v2. - DOI
-
- Arslan A. et al. (2022) Analysis of structural variation among inbred mouse strains identifies genetic factors for autism-related traits. BioRxiv. https://www.biorxiv.org/content/10.1101/2021.02.18.431863v1. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
