

CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting
- PMID: 35609983
- PMCID: PMC9252749
- DOI: 10.1093/nar/gkac394
CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting
Abstract
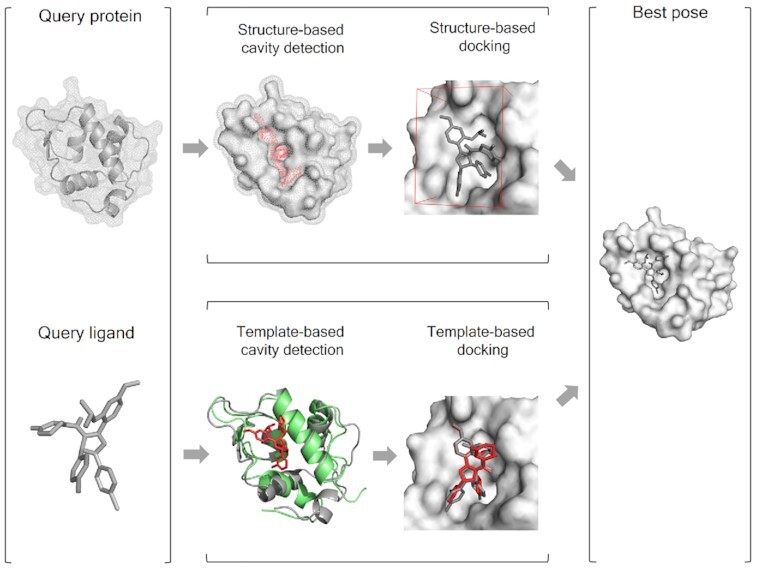
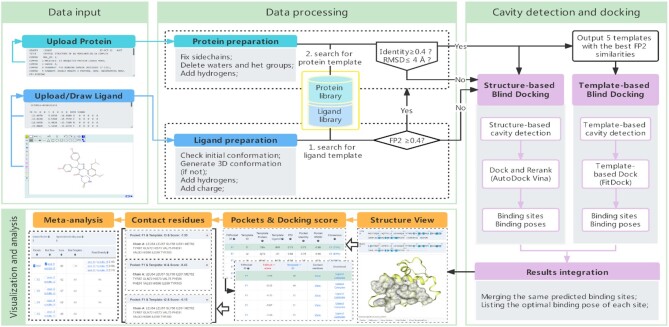
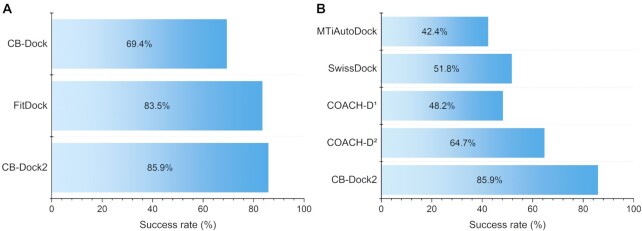
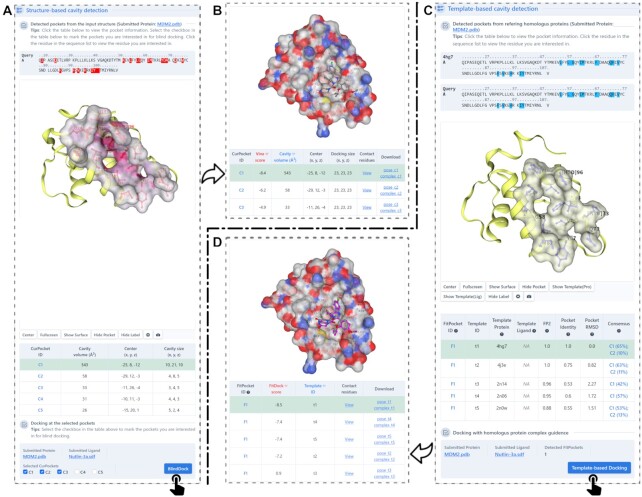
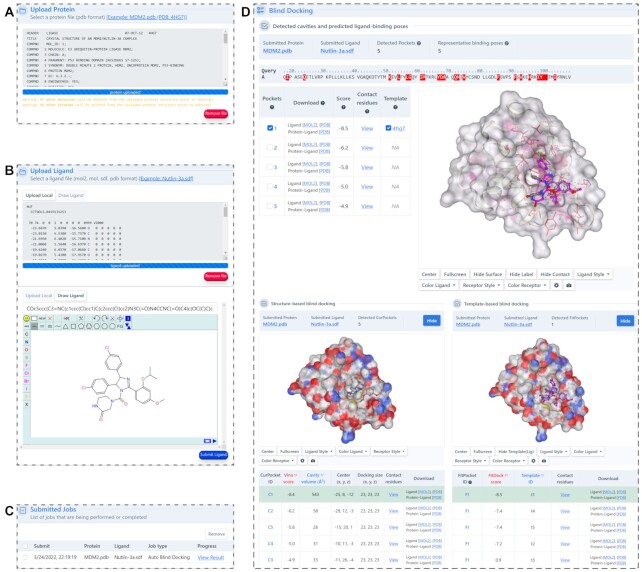
Protein-ligand blind docking is a powerful method for exploring the binding sites of receptors and the corresponding binding poses of ligands. It has seen wide applications in pharmaceutical and biological researches. Previously, we proposed a blind docking server, CB-Dock, which has been under heavy use (over 200 submissions per day) by researchers worldwide since 2019. Here, we substantially improved the docking method by combining CB-Dock with our template-based docking engine to enhance the accuracy in binding site identification and binding pose prediction. In the benchmark tests, it yielded the success rate of ∼85% for binding pose prediction (RMSD < 2.0 Å), which outperformed original CB-Dock and most popular blind docking tools. This updated docking server, named CB-Dock2, reconfigured the input and output web interfaces, together with a highly automatic docking pipeline, making it a particularly efficient and easy-to-use tool for the bioinformatics and cheminformatics communities. The web server is freely available at https://cadd.labshare.cn/cb-dock2/.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
