

Predicting cancer prognosis and drug response from the tumor microbiome
- PMID: 35610202
- PMCID: PMC9130323
- DOI: 10.1038/s41467-022-30512-3
Predicting cancer prognosis and drug response from the tumor microbiome
Retraction in
-
Retraction Note: Predicting cancer prognosis and drug response from the tumor microbiome.Nat Commun. 2025 Sep 17;16(1):8308. doi: 10.1038/s41467-025-64170-y. Nat Commun. 2025. PMID: 40962839 Free PMC article. No abstract available.
Abstract
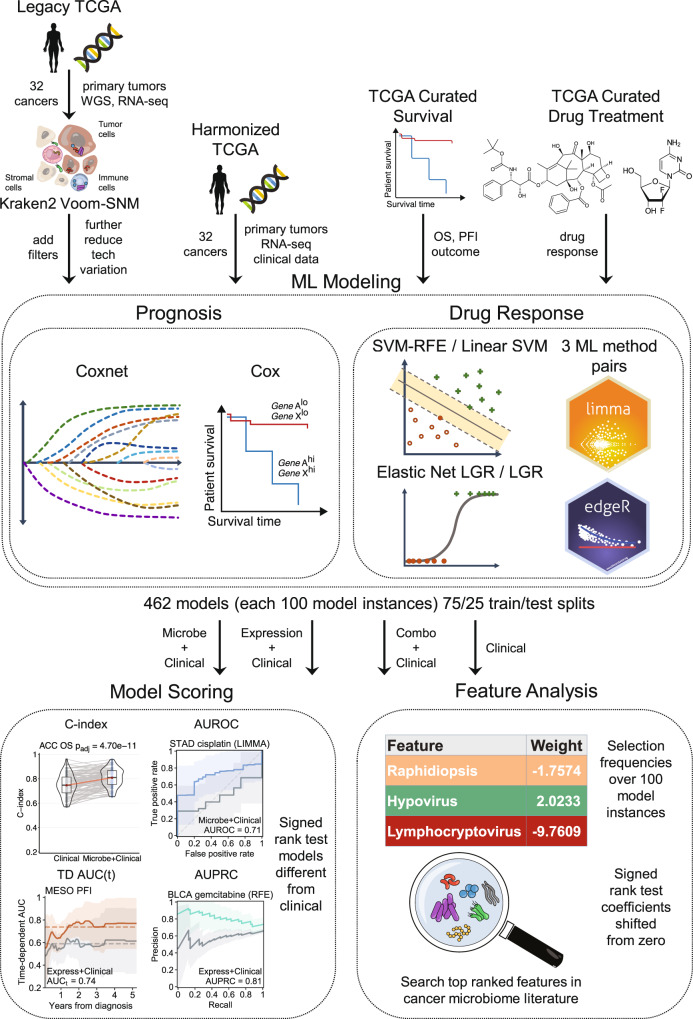
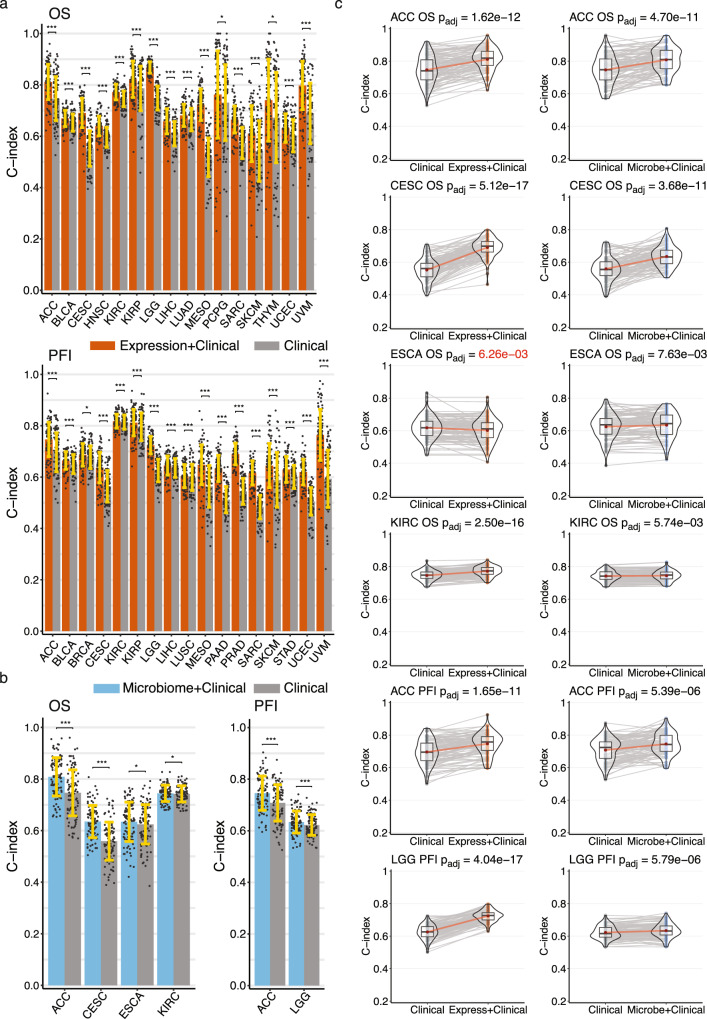
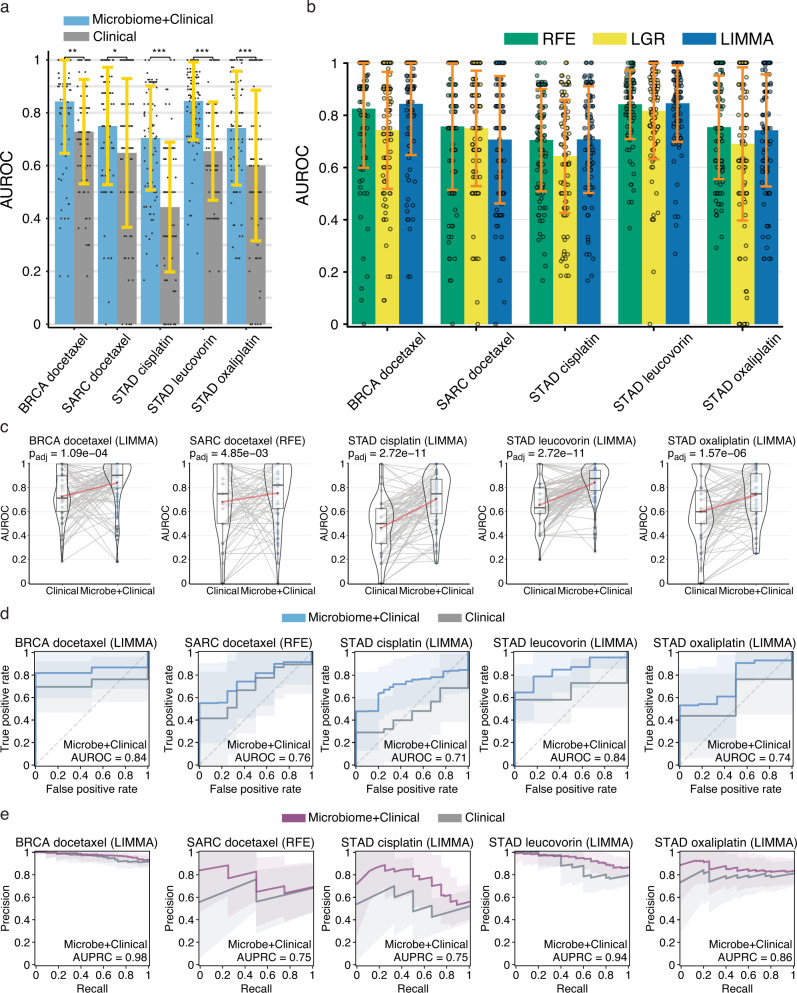
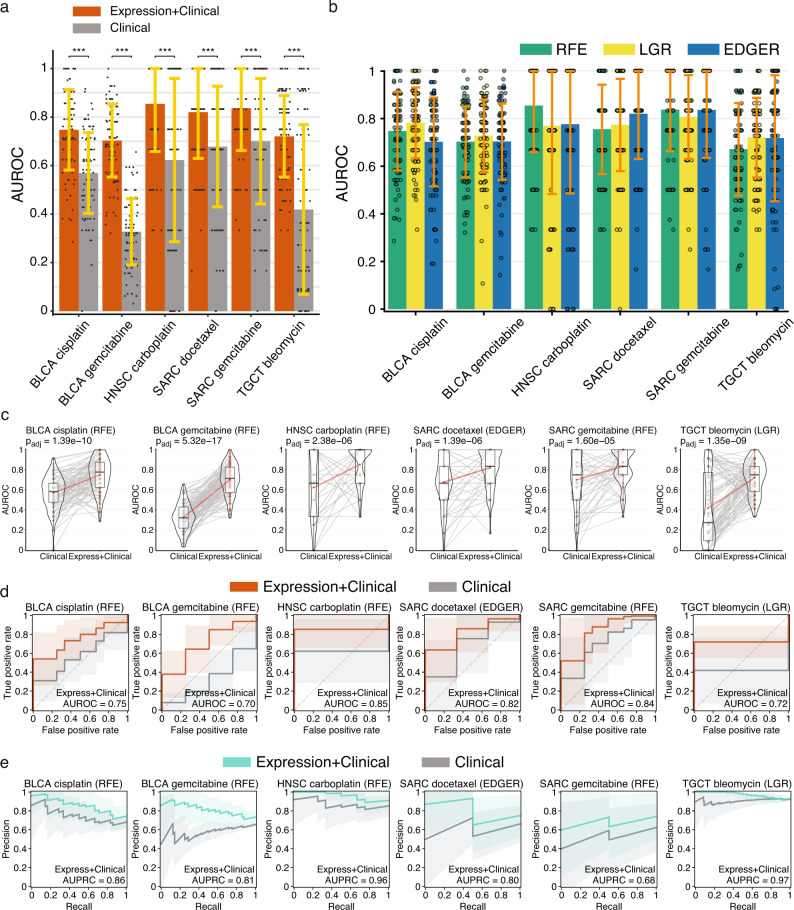
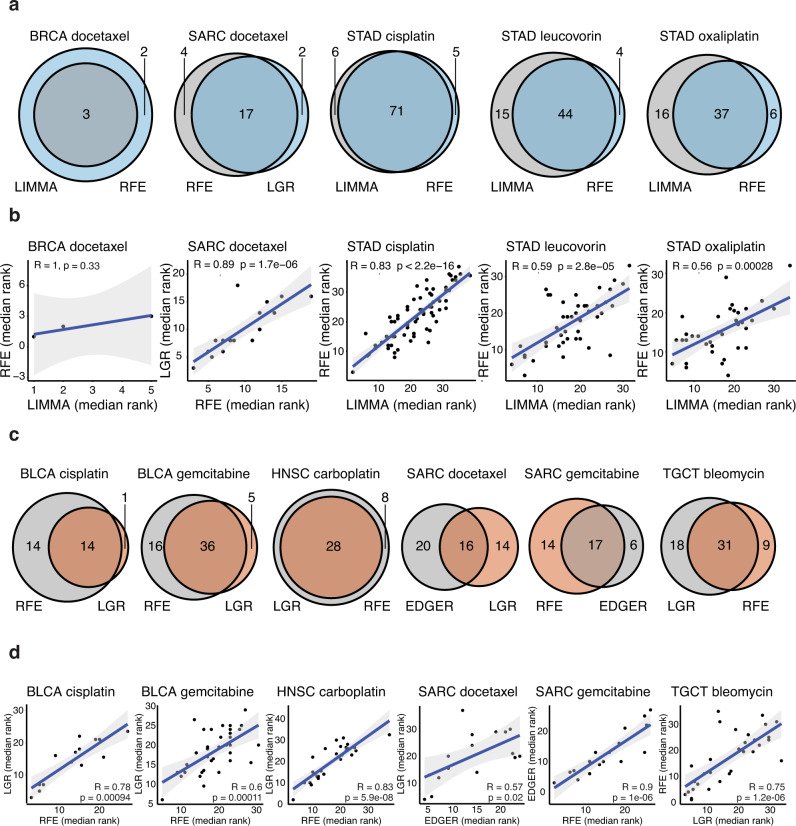
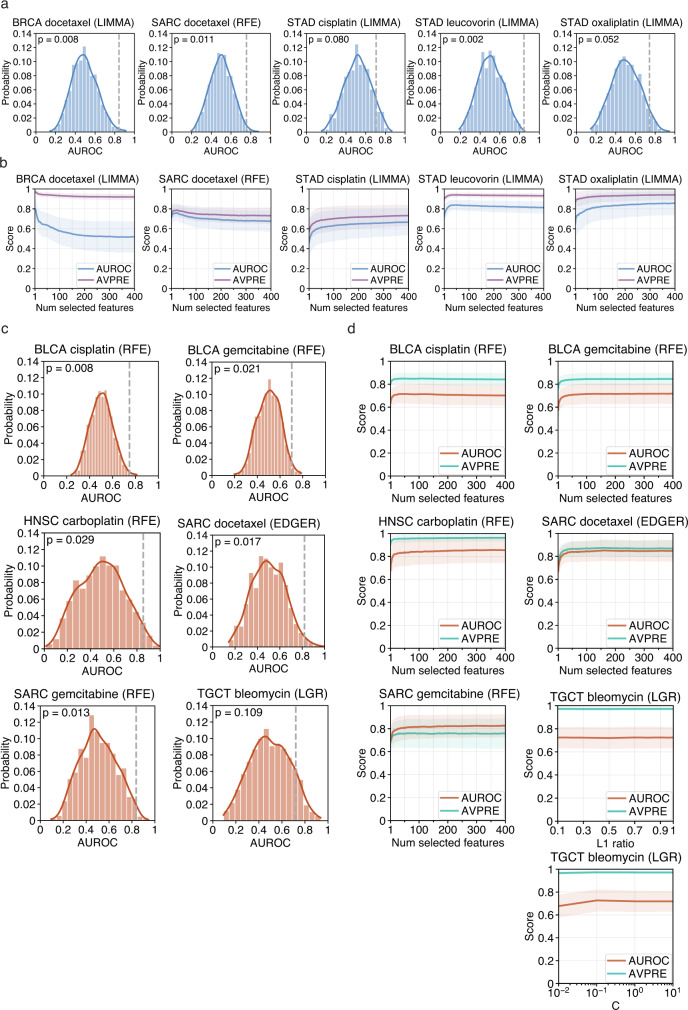
Tumor gene expression is predictive of patient prognosis in some cancers. However, RNA-seq and whole genome sequencing data contain not only reads from host tumor and normal tissue, but also reads from the tumor microbiome, which can be used to infer the microbial abundances in each tumor. Here, we show that tumor microbial abundances, alone or in combination with tumor gene expression, can predict cancer prognosis and drug response to some extent-microbial abundances are significantly less predictive of prognosis than gene expression, although similarly as predictive of drug response, but in mostly different cancer-drug combinations. Thus, it appears possible to leverage existing sequencing technology, or develop new protocols, to obtain more non-redundant information about prognosis and drug response from RNA-seq and whole genome sequencing experiments than could be obtained from tumor gene expression or genomic data alone.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Ahluwalia, P., Kolhe, R. & Gahlay, G. K. The clinical relevance of gene expression based prognostic signatures in colorectal cancer. Biochimica et. Biophysica Acta (BBA)Rev. Cancer1875, 188513 (2021). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
