

Characterising genome architectures using genome decomposition analysis
- PMID: 35610562
- PMCID: PMC9131526
- DOI: 10.1186/s12864-022-08616-3
Characterising genome architectures using genome decomposition analysis
Abstract
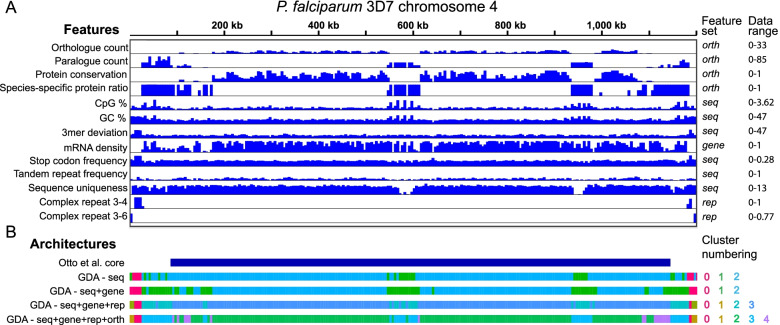
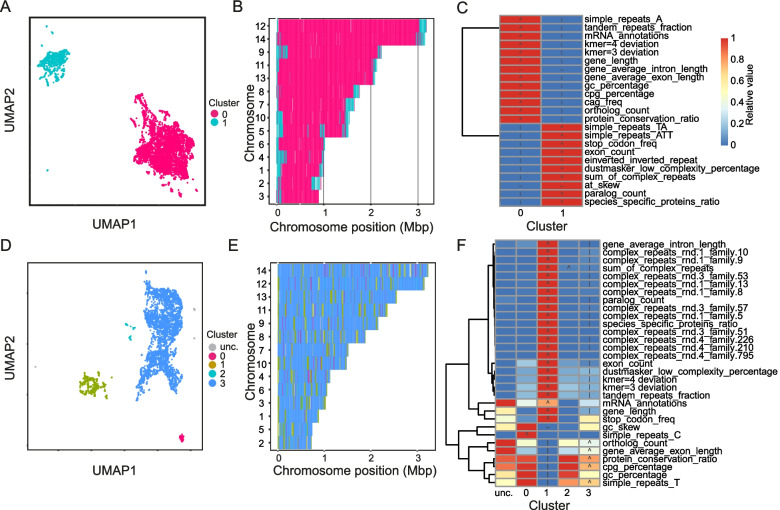
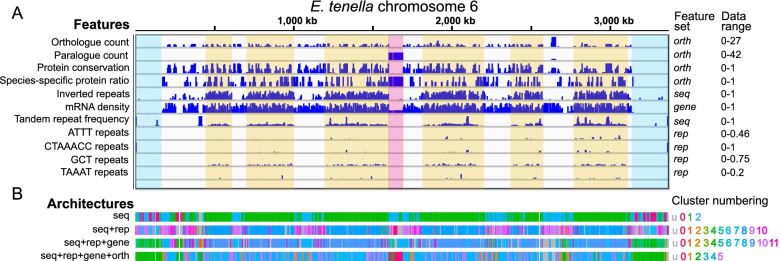
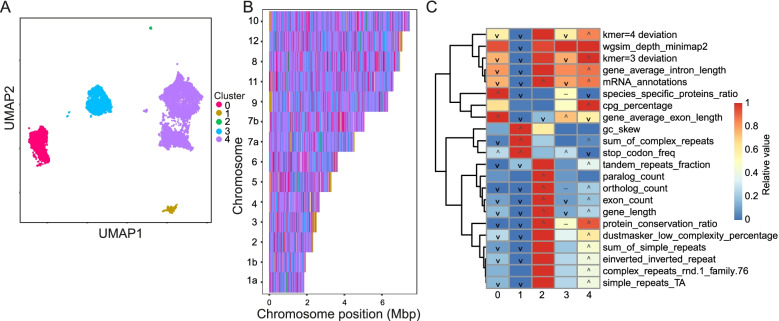
Genome architecture describes how genes and other features are arranged in genomes. These arrangements reflect the evolutionary pressures on genomes and underlie biological processes such as chromosomal segregation and the regulation of gene expression. We present a new tool called Genome Decomposition Analysis (GDA) that characterises genome architectures and acts as an accessible approach for discovering hidden features of a genome assembly. With the imminent deluge of high-quality genome assemblies from projects such as the Darwin Tree of Life and the Earth BioGenome Project, GDA has been designed to facilitate their exploration and the discovery of novel genome biology. We highlight the effectiveness of our approach in characterising the genome architectures of single-celled eukaryotic parasites from the phylum Apicomplexa and show that it scales well to large genomes.
Keywords: Apicomplexa; Chromosome structure; Genome architecture; Genome assembly; Parasites; Plasmodium.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
