

Identification of vaccine targets & design of vaccine against SARS-CoV-2 coronavirus using computational and deep learning-based approaches
- PMID: 35611169
- PMCID: PMC9124463
- DOI: 10.7717/peerj.13380
Identification of vaccine targets & design of vaccine against SARS-CoV-2 coronavirus using computational and deep learning-based approaches
Abstract
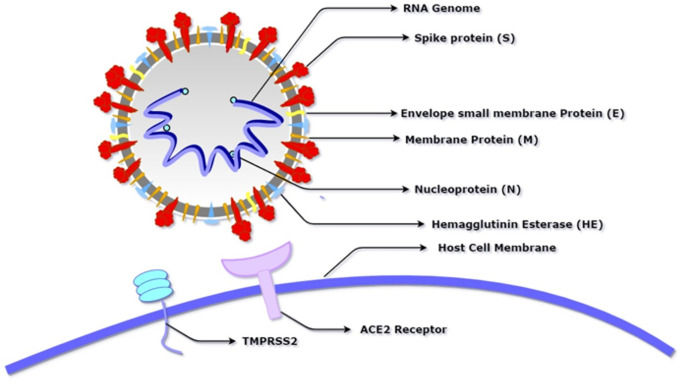
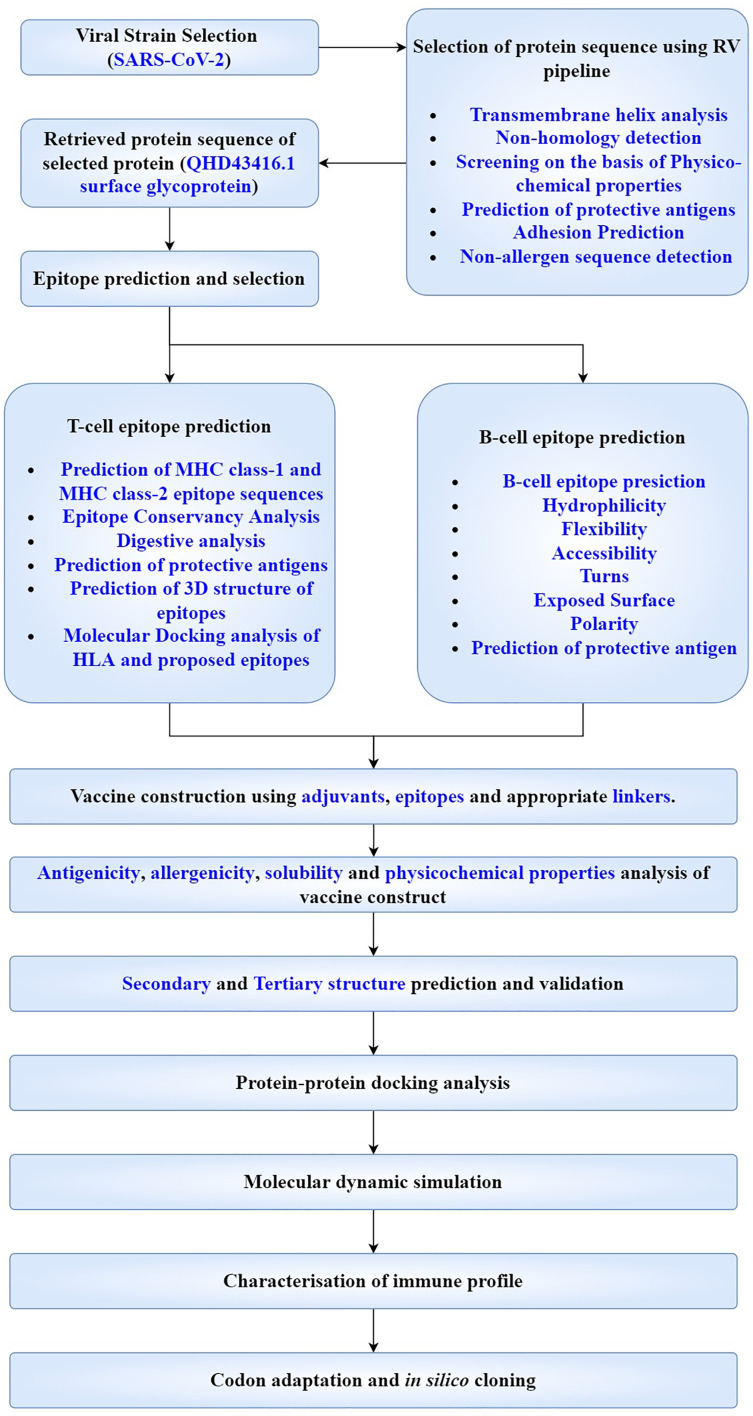
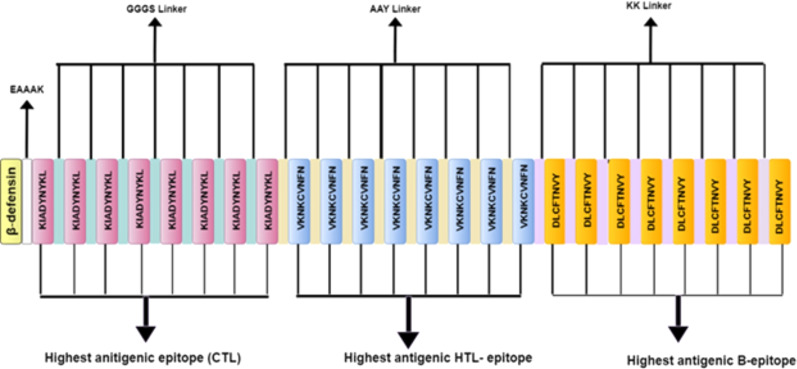
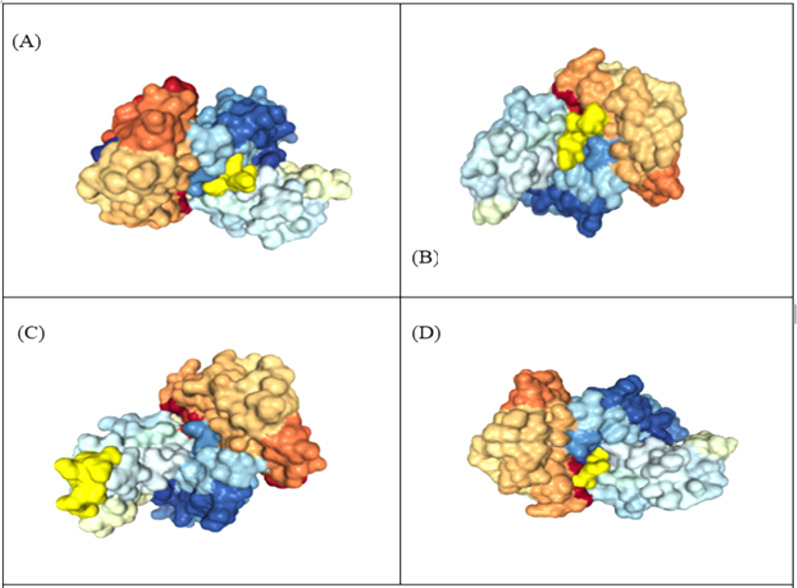
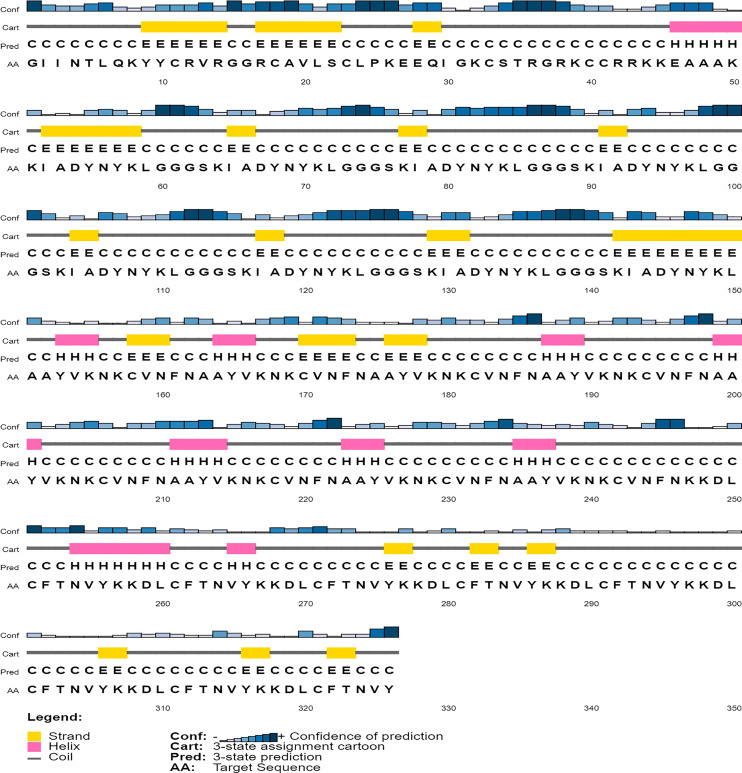
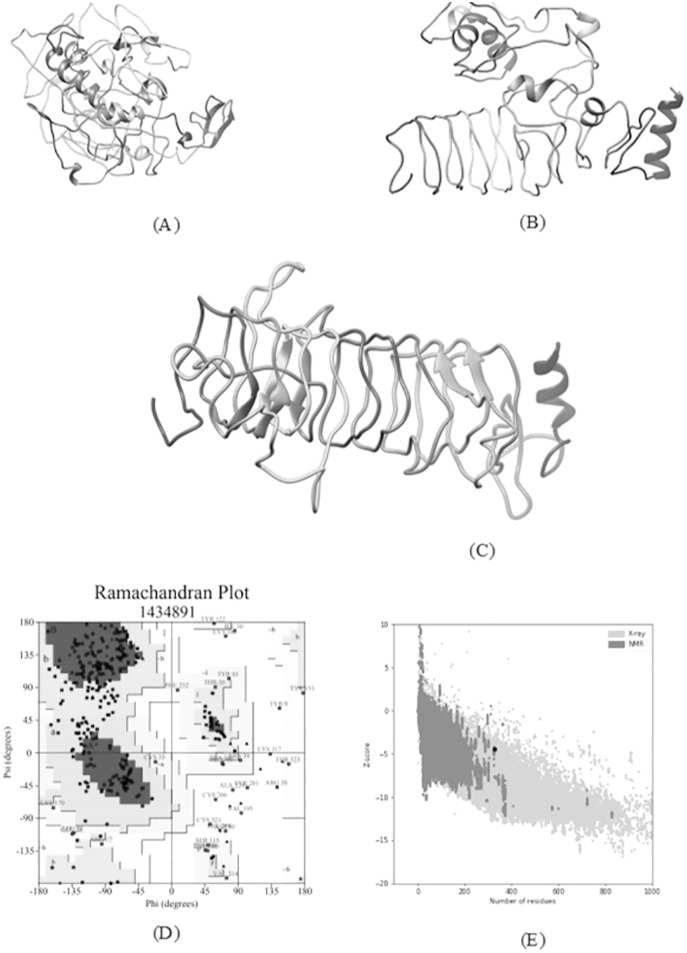
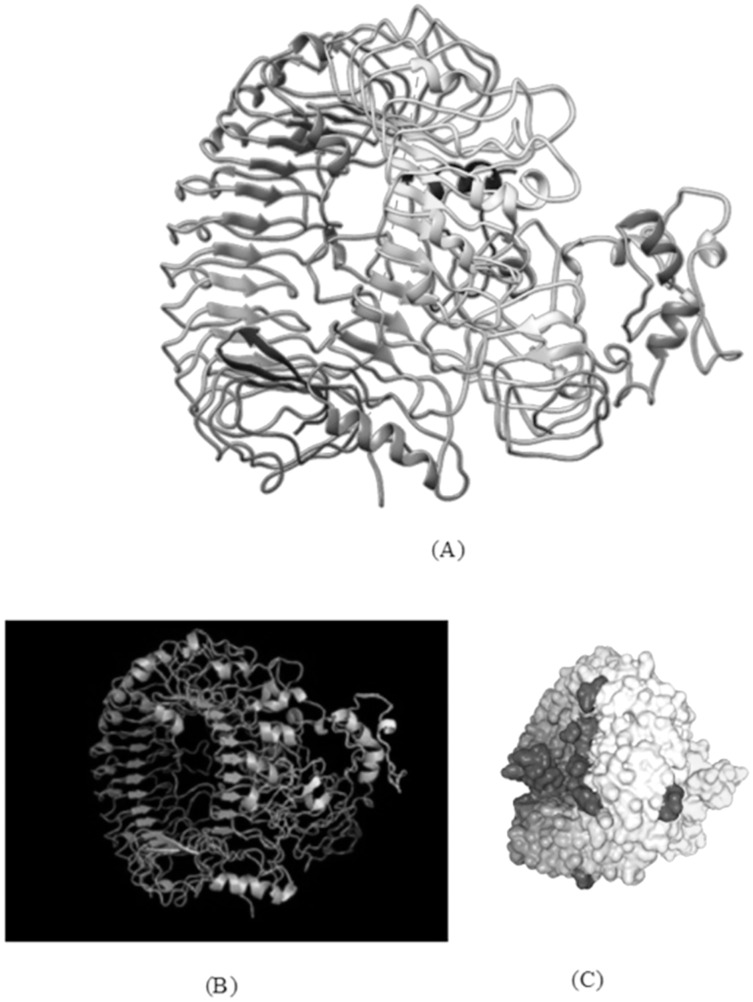
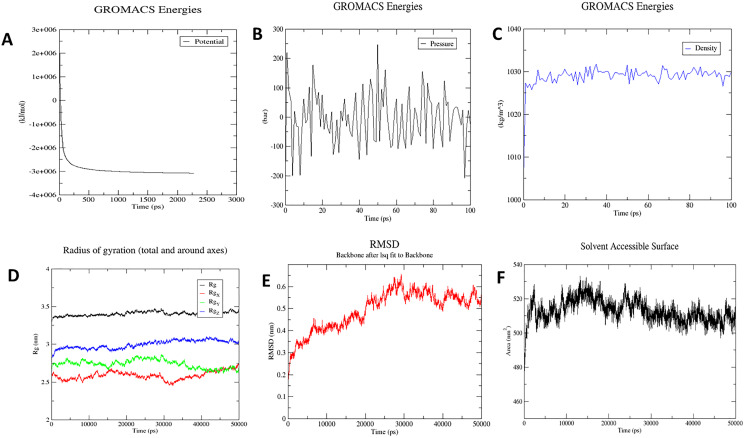
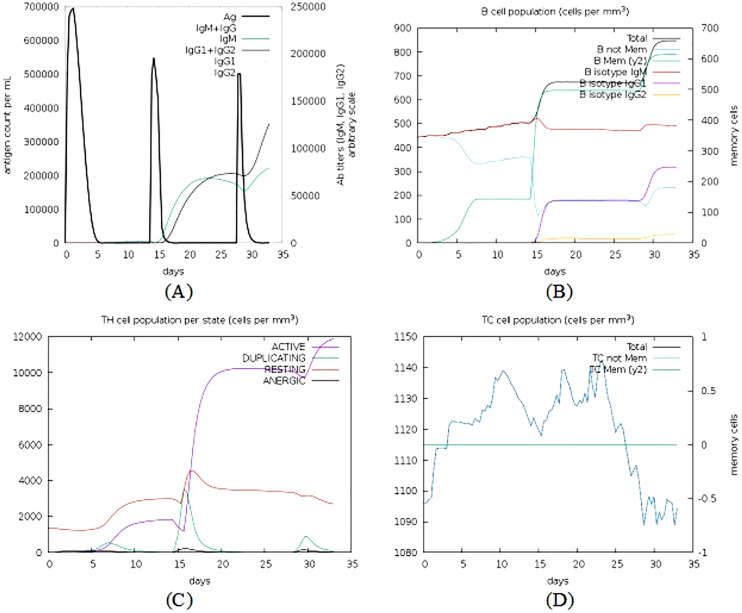
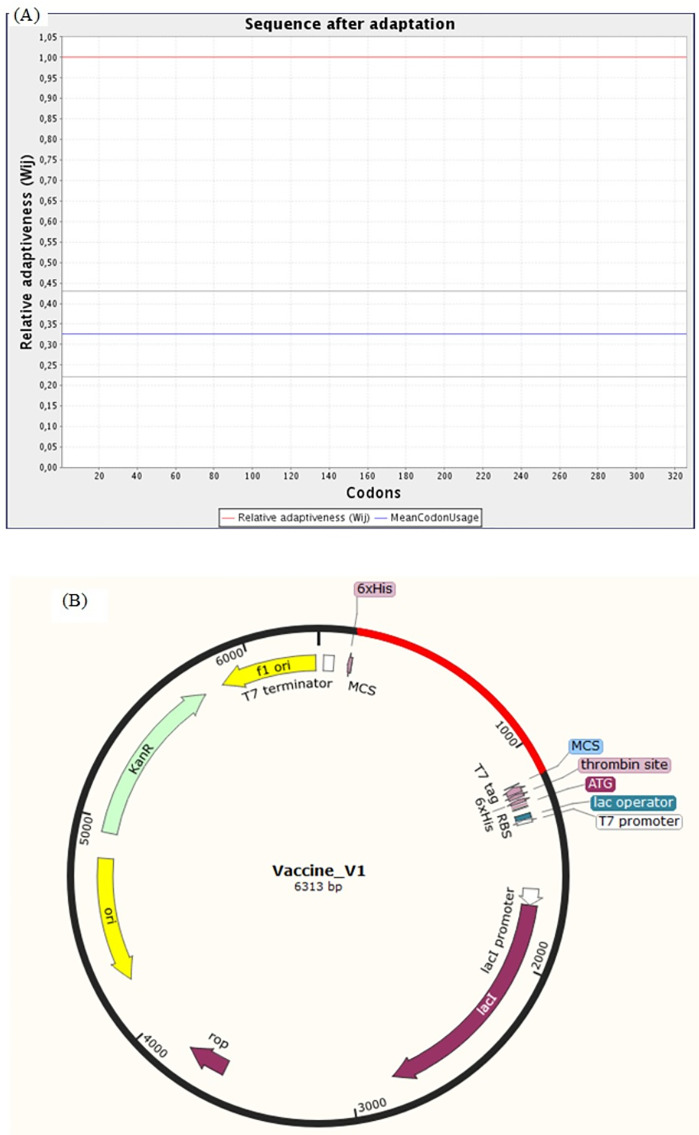
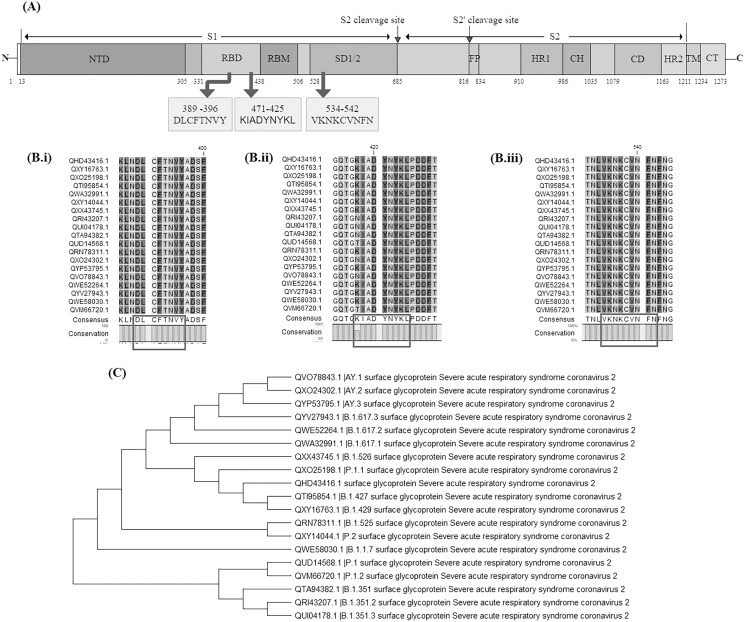
An unusual pneumonia infection, named COVID-19, was reported on December 2019 in China. It was reported to be caused by a novel coronavirus which has infected approximately 220 million people worldwide with a death toll of 4.5 million as of September 2021. This study is focused on finding potential vaccine candidates and designing an in-silico subunit multi-epitope vaccine candidates using a unique computational pipeline, integrating reverse vaccinology, molecular docking and simulation methods. A protein named spike protein of SARS-CoV-2 with the GenBank ID QHD43416.1 was shortlisted as a potential vaccine candidate and was examined for presence of B-cell and T-cell epitopes. We also investigated antigenicity and interaction with distinct polymorphic alleles of the epitopes. High ranking epitopes such as DLCFTNVY (B cell epitope), KIADYNKL (MHC Class-I) and VKNKCVNFN (MHC class-II) were shortlisted for subsequent analysis. Digestion analysis verified the safety and stability of the shortlisted peptides. Docking study reported a strong binding of proposed peptides with HLA-A*02 and HLA-B7 alleles. We used standard methods to construct vaccine model and this construct was evaluated further for its antigenicity, physicochemical properties, 2D and 3D structure prediction and validation. Further, molecular docking followed by molecular dynamics simulation was performed to evaluate the binding affinity and stability of TLR-4 and vaccine complex. Finally, the vaccine construct was reverse transcribed and adapted for E. coli strain K 12 prior to the insertion within the pET-28-a (+) vector for determining translational and microbial expression followed by conservancy analysis. Also, six multi-epitope subunit vaccines were constructed using different strategies containing immunogenic epitopes, appropriate adjuvants and linker sequences. We propose that our vaccine constructs can be used for downstream investigations using in-vitro and in-vivo studies to design effective and safe vaccine against different strains of COVID-19.
Keywords: Deep learning; Epitopes; Molecular docking; Reverse vaccinology; SARS-CoV-2; Vaccine-designing.
© 2022 Abbasi et al.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, Bourexis D, Brister JR, Bryant SH, Canese K, Cavanaugh M, Charowhas C, Clark K, Dondoshansky I, Feolo M, Fitzpatrick L, Funk K, Geer LY, Gorelenkov V, Graeff A, Hlavina W, Holmes B, Johnson M, Kattman B, Khotomlianski V, Kimchi A, Kimelman M, Kimura M, Kitts P, Klimke W, Kotliarov A, Krasnov S, Kuznetsov A, Landrum MJ, Landsman D, Lathrop S, Lee JM, Leubsdorf C, Lu Z, Madden TL, Marchler-Bauer A, Malheiro A, Meric P, Karsch-Mizrachi I, Mnev A, Murphy T, Orris R, Ostell J, O’Sullivan C, Palanigobu V, Panchenko AR, Phan L, Pierov B, Pruitt KD, Rodarmer K, Sayers EW, Schneider V, Schoch CL, Schuler GD, Sherry ST, Siyan K, Soboleva A, Soussov V, Starchenko G, Tatusova TA, Thibaud-Nissen F, Todorov K, Trawick BW, Vakatov D, Ward M, Yaschenko E, Zasypkin A, Zbicz K. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 2018;46(D1):D8–D13. doi: 10.1093/nar/gkx1095. - DOI - PMC - PubMed
-
- Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Scientific Reports. 2017;7(1):9232. doi: 10.1038/s41598-017-09199-w. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
