

Targeting Adiponectin Receptor 1 Phosphorylation Against Ischemic Heart Failure
- PMID: 35611695
- PMCID: PMC9308652
- DOI: 10.1161/CIRCRESAHA.121.319976
Targeting Adiponectin Receptor 1 Phosphorylation Against Ischemic Heart Failure
Abstract
Background: Despite significantly reduced acute myocardial infarction (MI) mortality in recent years, ischemic heart failure continues to escalate. Therapeutic interventions effectively reversing pathological remodeling are an urgent unmet medical need. We recently demonstrated that AdipoR1 (APN [adiponectin] receptor 1) phosphorylation by GRK2 (G-protein-coupled receptor kinase 2) contributes to maladaptive remodeling in the ischemic heart. The current study clarified the underlying mechanisms leading to AdipoR1 phosphorylative desensitization and investigated whether blocking AdipoR1 phosphorylation may restore its protective signaling, reversing post-MI remodeling.
Methods: Specific sites and underlying molecular mechanisms responsible for AdipoR1 phosphorylative desensitization were investigated in vitro (neonatal and adult cardiomyocytes). The effects of AdipoR1 phosphorylation inhibition upon APN post-MI remodeling and heart failure progression were investigated in vivo.
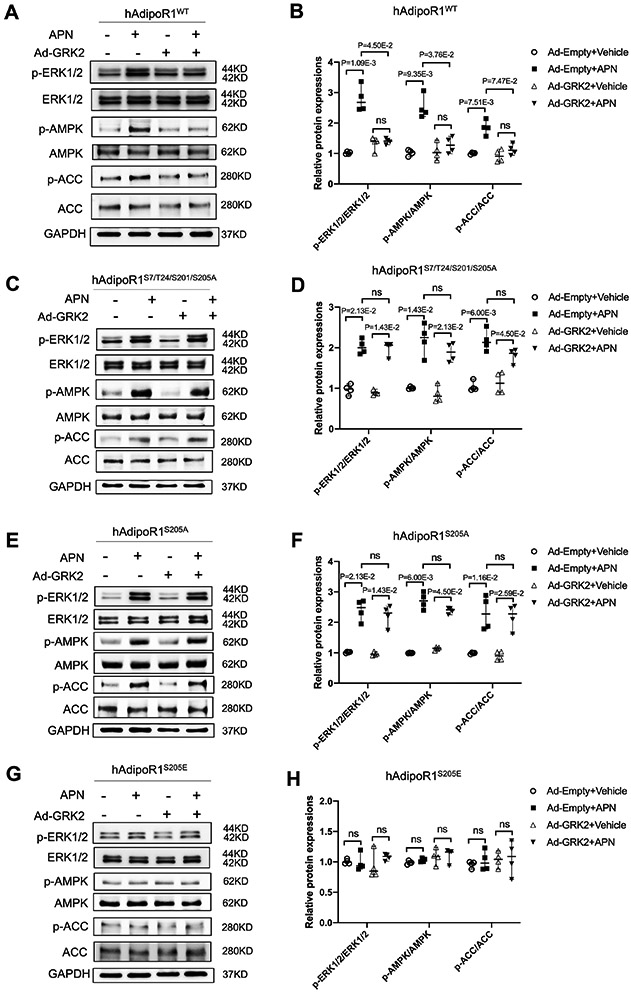
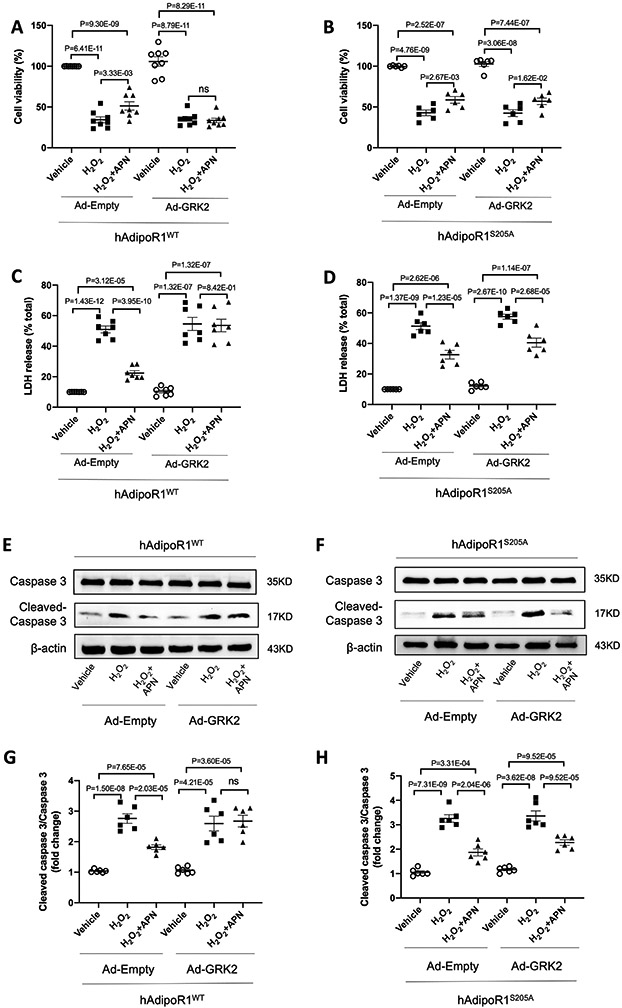
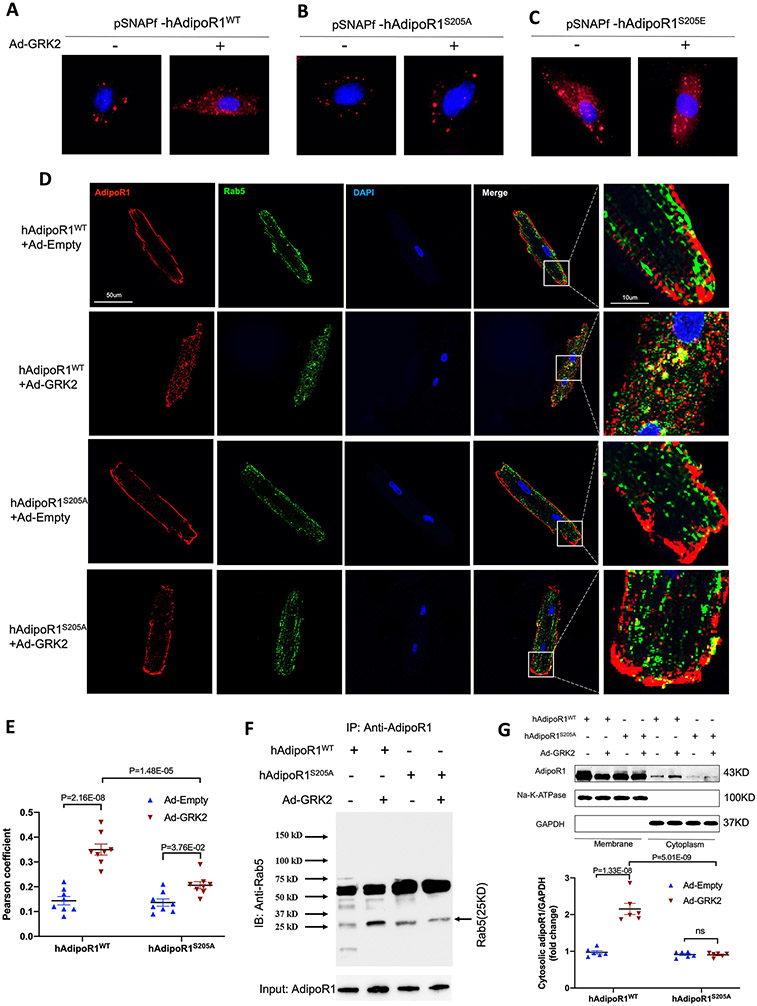
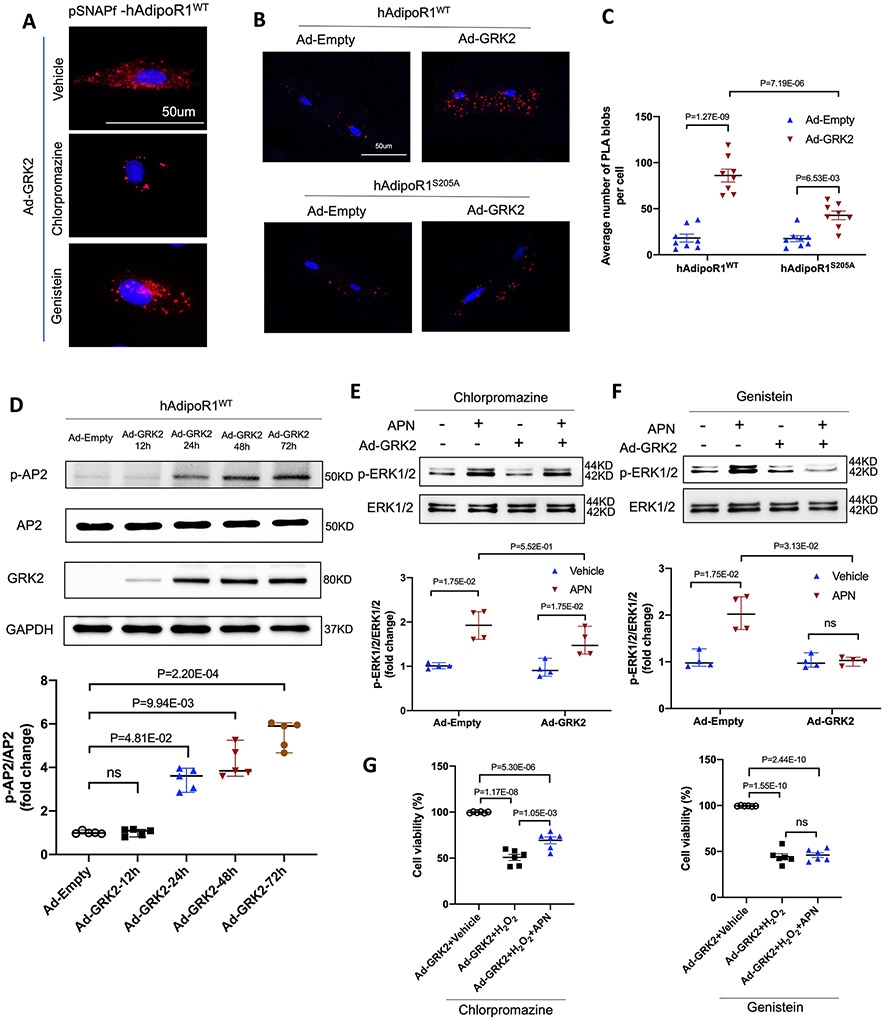
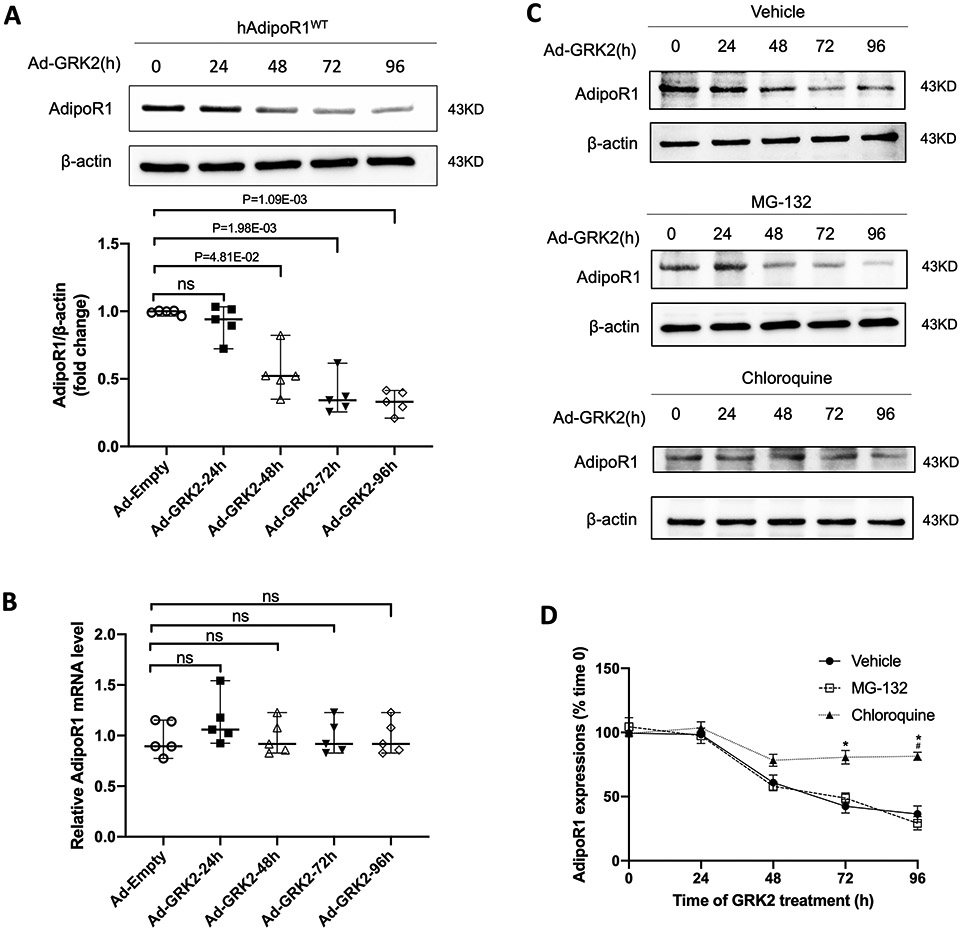
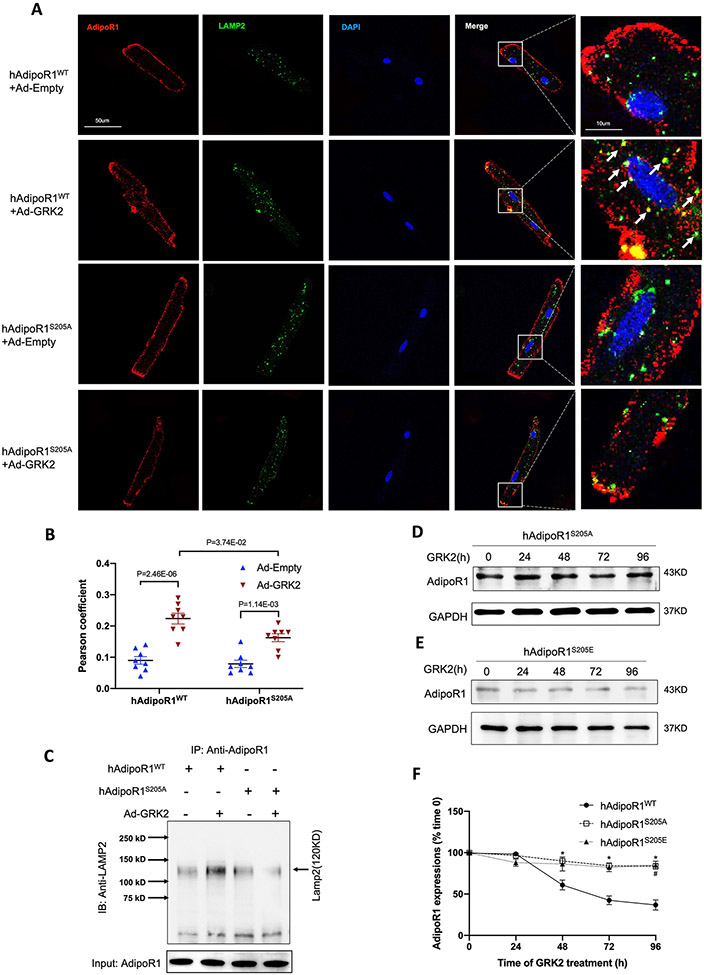
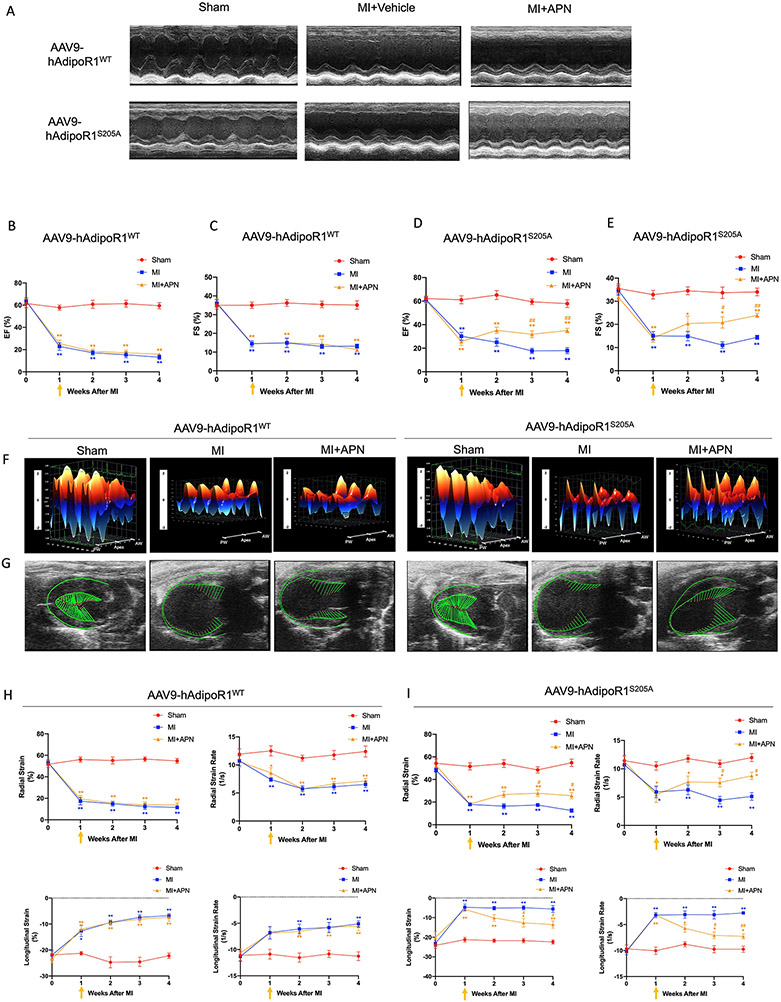
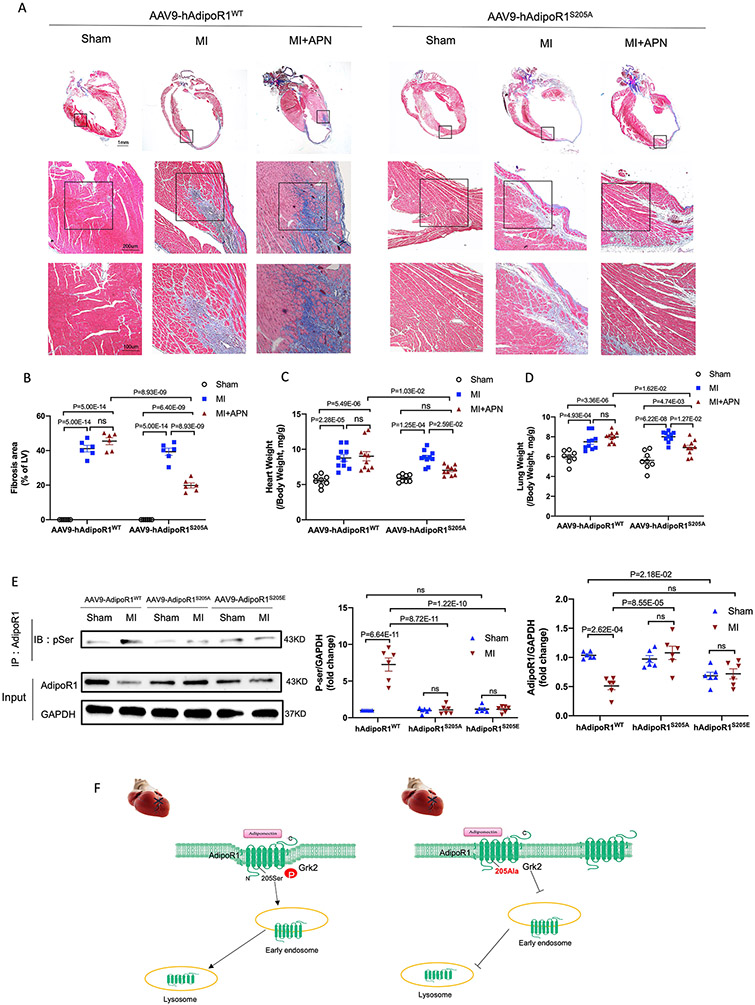
Results: Among 4 previously identified sites sensitive to GRK2 phosphorylation, alanine substitution of Ser205 (AdipoR1S205A), but not other 3 sites, rescued GRK2-suppressed AdipoR1 functions, restoring APN-induced cell salvage kinase activation and reducing oxidative cell death. The molecular investigation followed by functional determination demonstrated that AdipoR1 phosphorylation promoted clathrin-dependent (not caveolae) endocytosis and lysosomal-mediated (not proteasome) degradation, reducing AdipoR1 protein level and suppressing AdipoR1-mediated cytoprotective action. GRK2-induced AdipoR1 endocytosis and degradation were blocked by AdipoR1S205A overexpression. Moreover, AdipoR1S205E (pseudophosphorylation) phenocopied GRK2 effects, promoted AdipoR1 endocytosis and degradation, and inhibited AdipoR1 biological function. Most importantly, AdipoR1 function was preserved during heart failure development in AdipoR1-KO (AdipoR1 knockout) mice reexpressing hAdipoR1S205A. APN administration in the failing heart reversed post-MI remodeling and improved cardiac function. However, reexpressing hAdipoR1WT in AdipoR1-KO mice failed to restore APN cardioprotection.
Conclusions: Ser205 is responsible for AdipoR1 phosphorylative desensitization in the failing heart. Blockade of AdipoR1 phosphorylation followed by pharmacological APN administration is a novel therapy effective in reversing post-MI remodeling and mitigating heart failure progression.
Keywords: adipokines; animals; cell death; endocytosis; heart failure.
Figures
Comment in
-
Leveraging Adipocyte-Cardiomyocyte Signaling to Treat Ischemic Heart Failure.Circ Res. 2022 Jul 8;131(2):165-167. doi: 10.1161/CIRCRESAHA.122.321392. Epub 2022 Jul 7. Circ Res. 2022. PMID: 35861740 No abstract available.
References
-
- Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. - PubMed
-
- Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;73:89–99. - PubMed
-
- Heusch G and Gersh BJ. Is Cardioprotection Salvageable? Circulation. 2020;141:415–417. - PubMed
-
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
