

Selective endocytosis of Ca2+-permeable AMPARs by the Alzheimer's disease risk factor CALM bidirectionally controls synaptic plasticity
- PMID: 35613266
- PMCID: PMC9132451
- DOI: 10.1126/sciadv.abl5032
Selective endocytosis of Ca2+-permeable AMPARs by the Alzheimer's disease risk factor CALM bidirectionally controls synaptic plasticity
Abstract
AMPA-type glutamate receptors (AMPARs) mediate fast excitatory neurotransmission, and the plastic modulation of their surface levels determines synaptic strength. AMPARs of different subunit compositions fulfill distinct roles in synaptic long-term potentiation (LTP) and depression (LTD) to enable learning. Largely unknown endocytic mechanisms mediate the subunit-selective regulation of the surface levels of GluA1-homomeric Ca2+-permeable (CP) versus heteromeric Ca2+-impermeable (CI) AMPARs. Here, we report that the Alzheimer's disease risk factor CALM controls the surface levels of CP-AMPARs and thereby reciprocally regulates LTP and LTD in vivo to modulate learning. We show that CALM selectively facilitates the endocytosis of ubiquitinated CP-AMPARs via a mechanism that depends on ubiquitin recognition by its ANTH domain but is independent of clathrin. Our data identify CALM and related ANTH domain-containing proteins as the core endocytic machinery that determines the surface levels of CP-AMPARs to bidirectionally control synaptic plasticity and modulate learning in the mammalian brain.
Figures
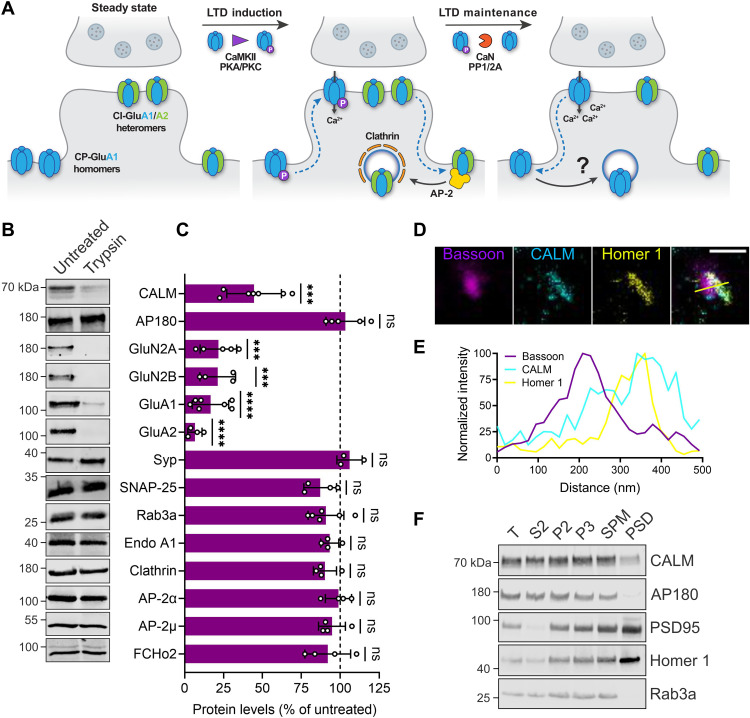
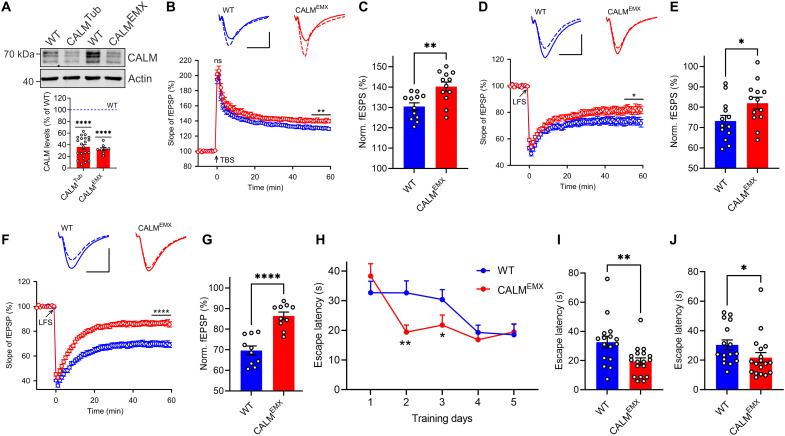
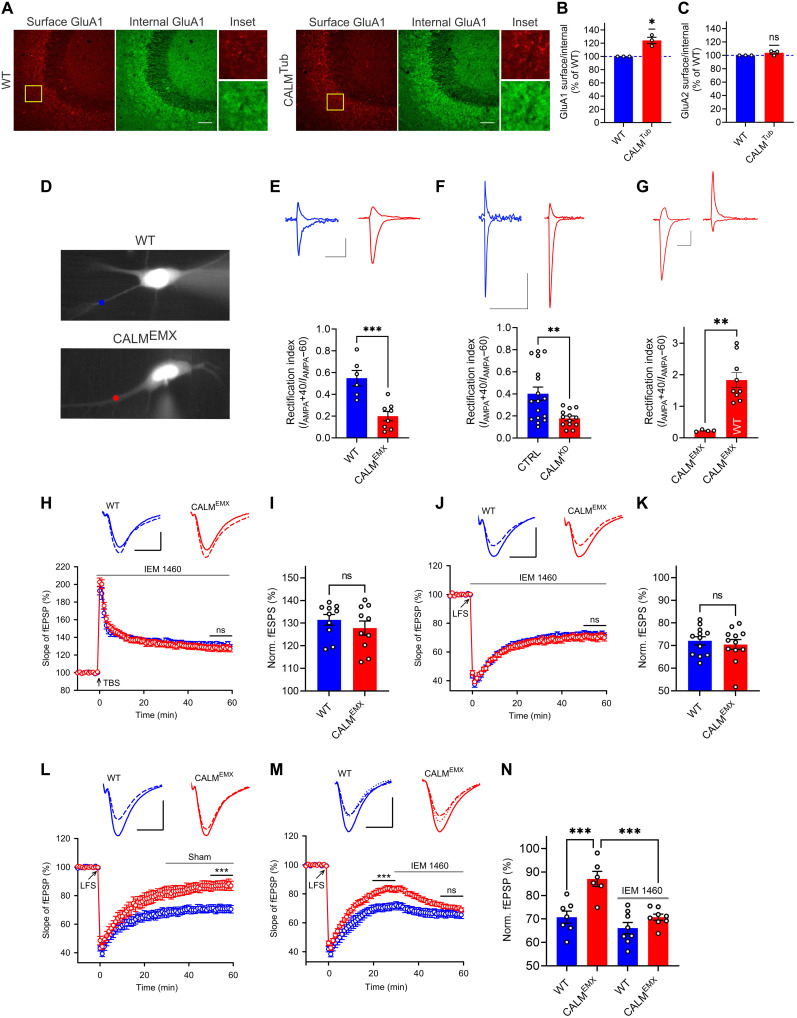
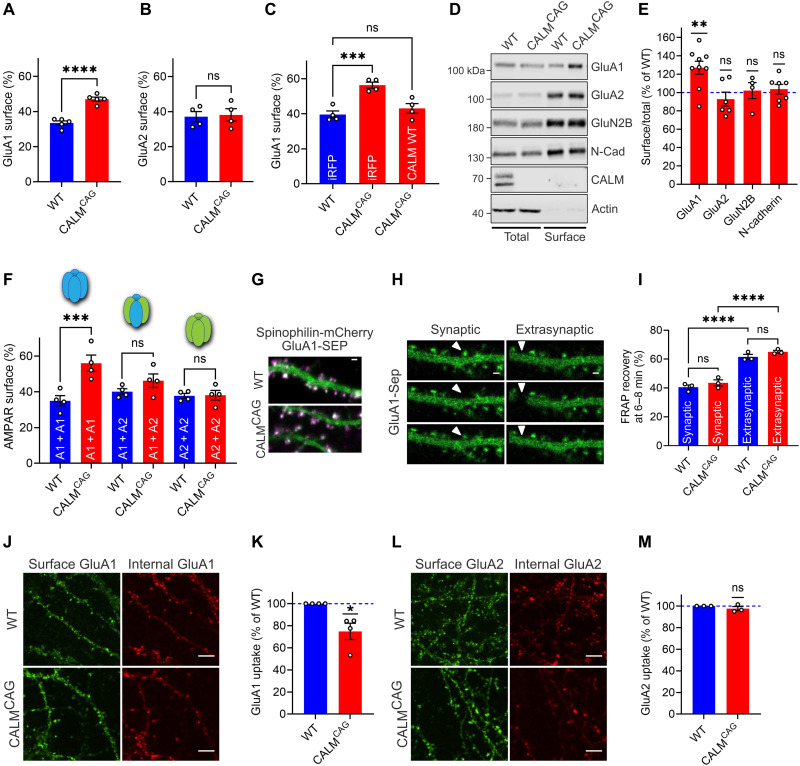
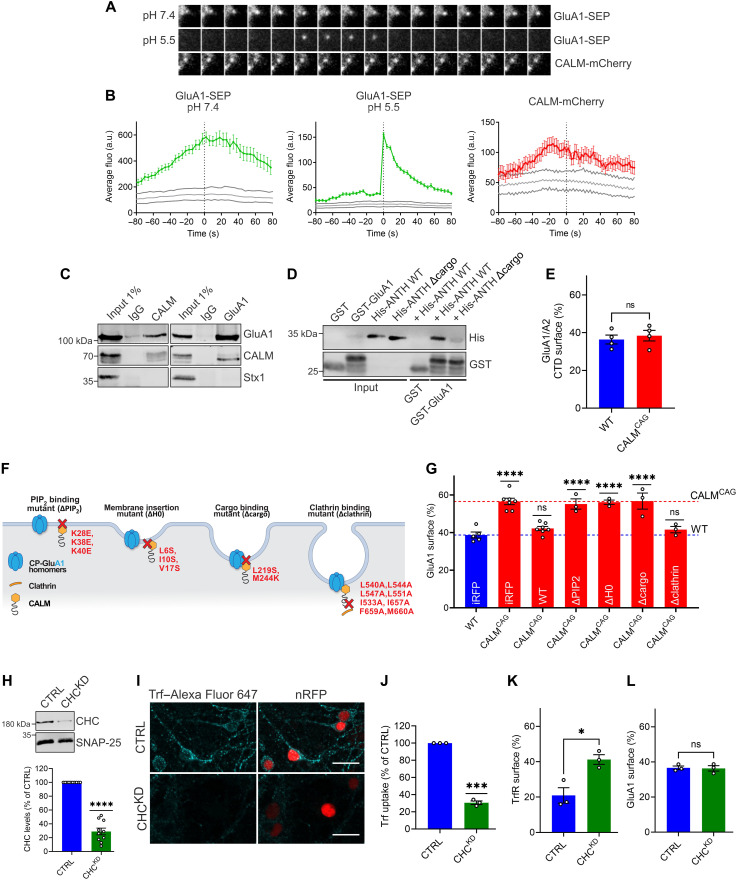
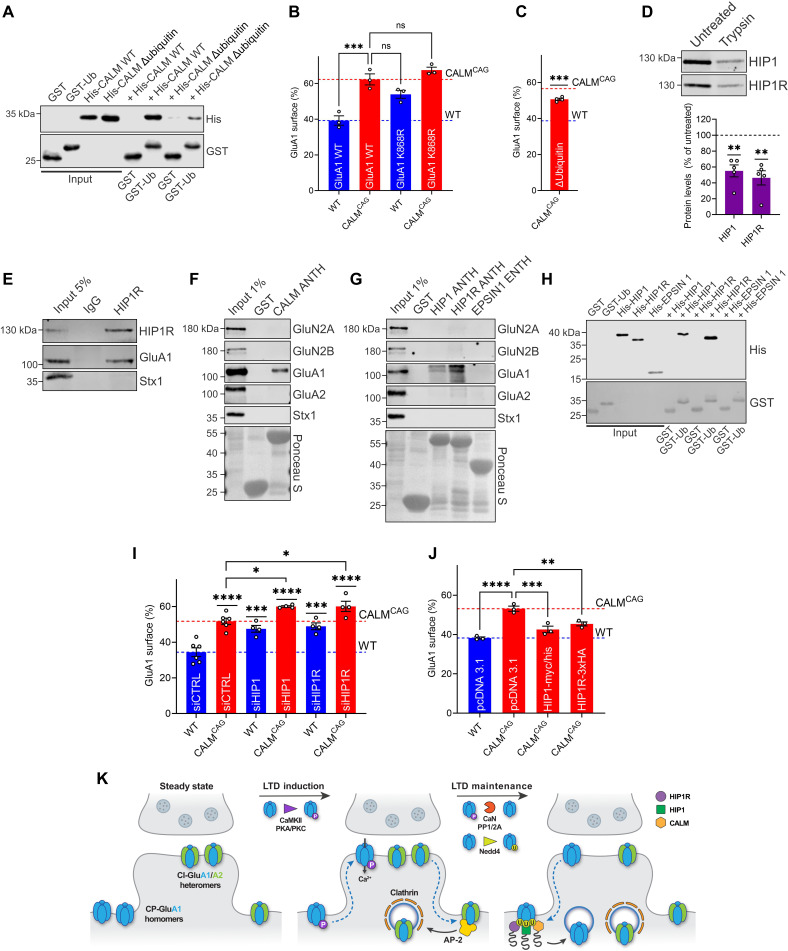
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
