

A vaccine targeting resistant tumours by dual T cell plus NK cell attack
- PMID: 35614223
- PMCID: PMC10253041
- DOI: 10.1038/s41586-022-04772-4
A vaccine targeting resistant tumours by dual T cell plus NK cell attack
Abstract
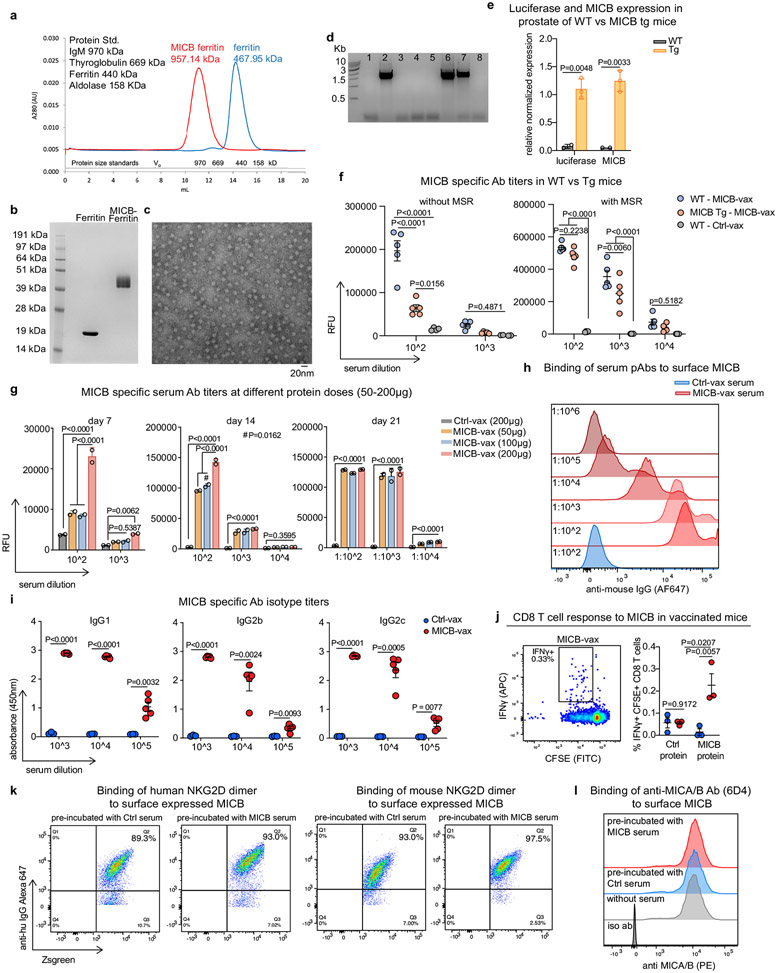
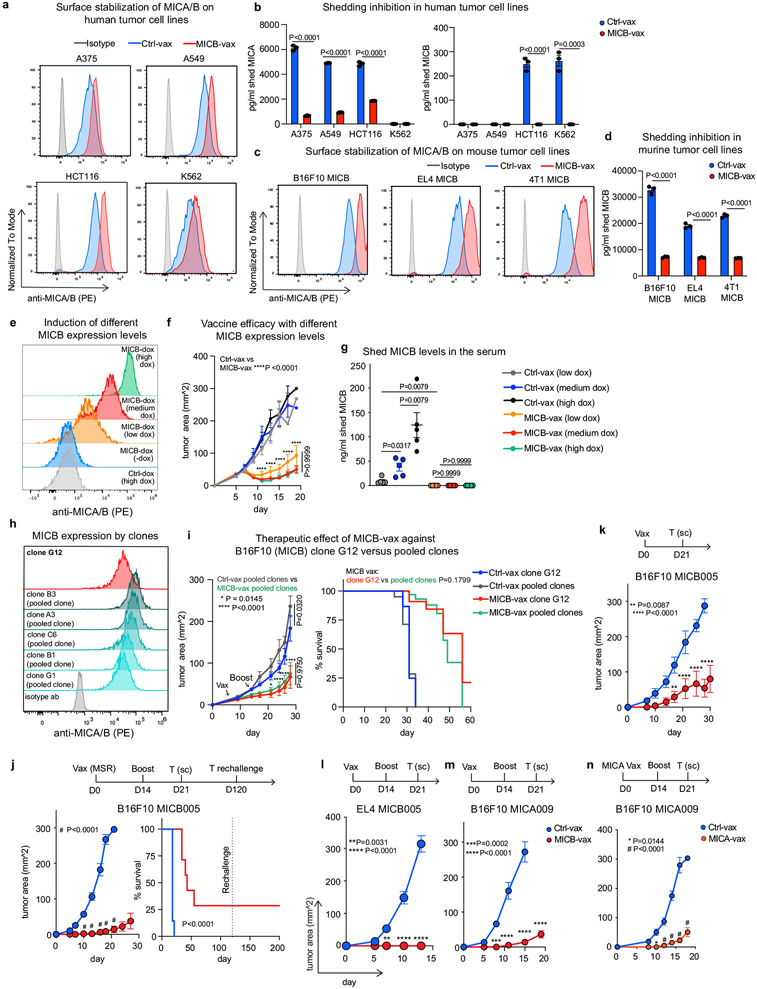
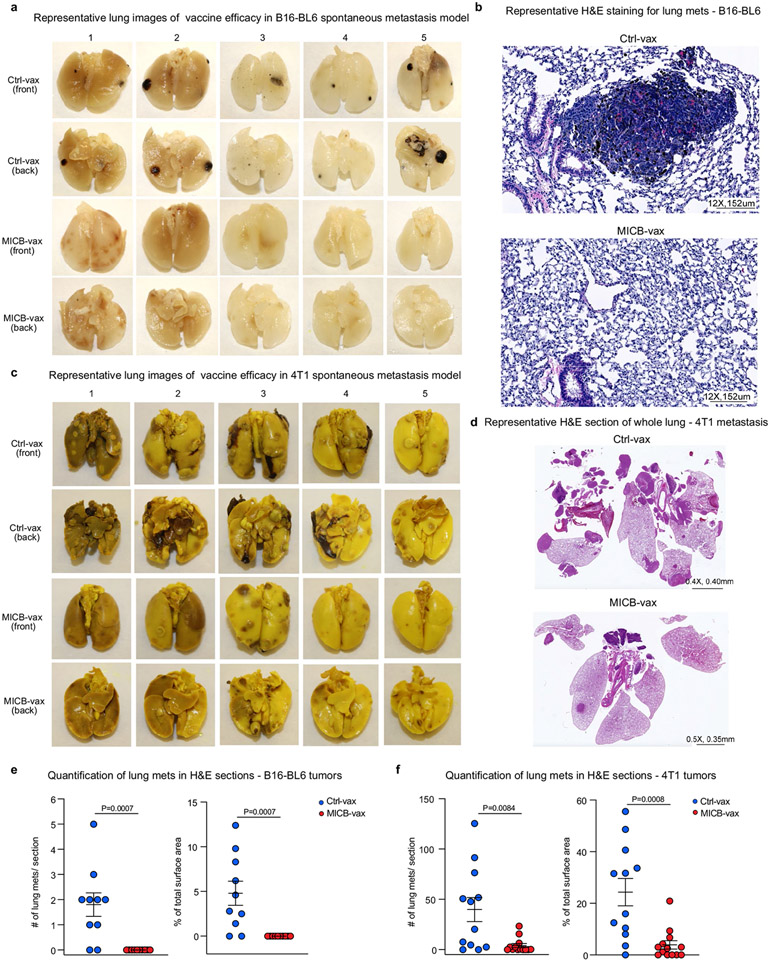
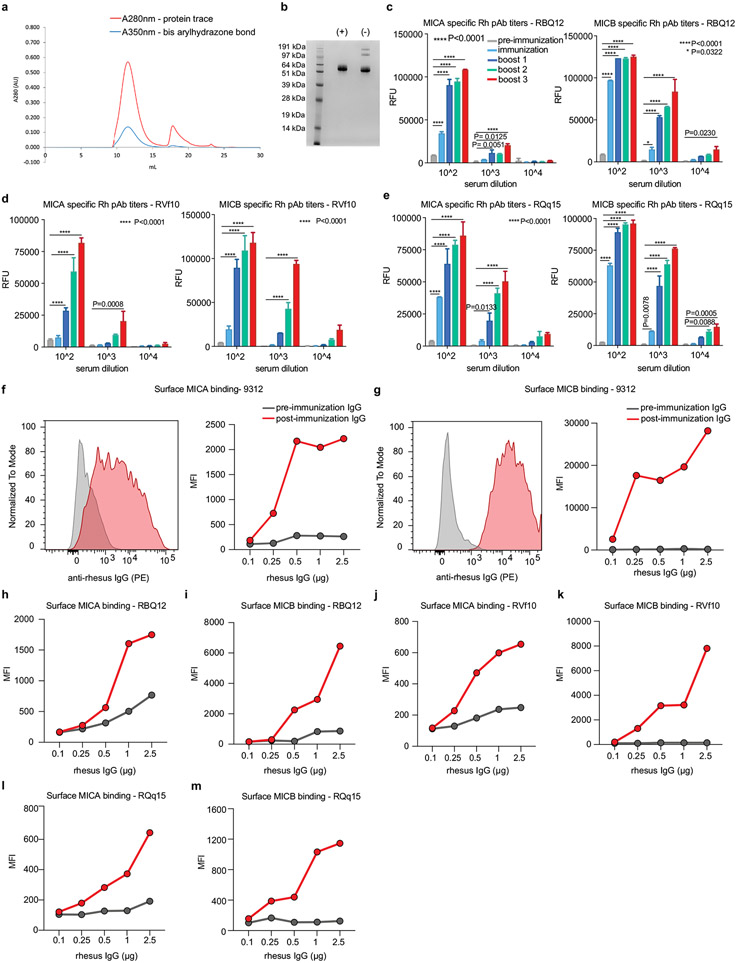
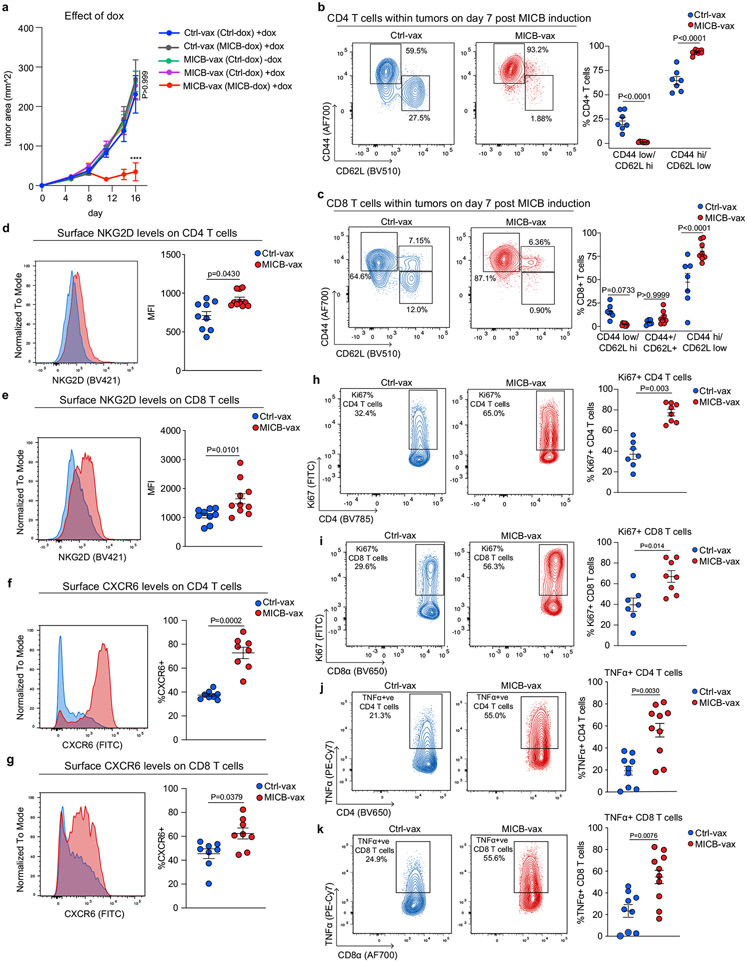
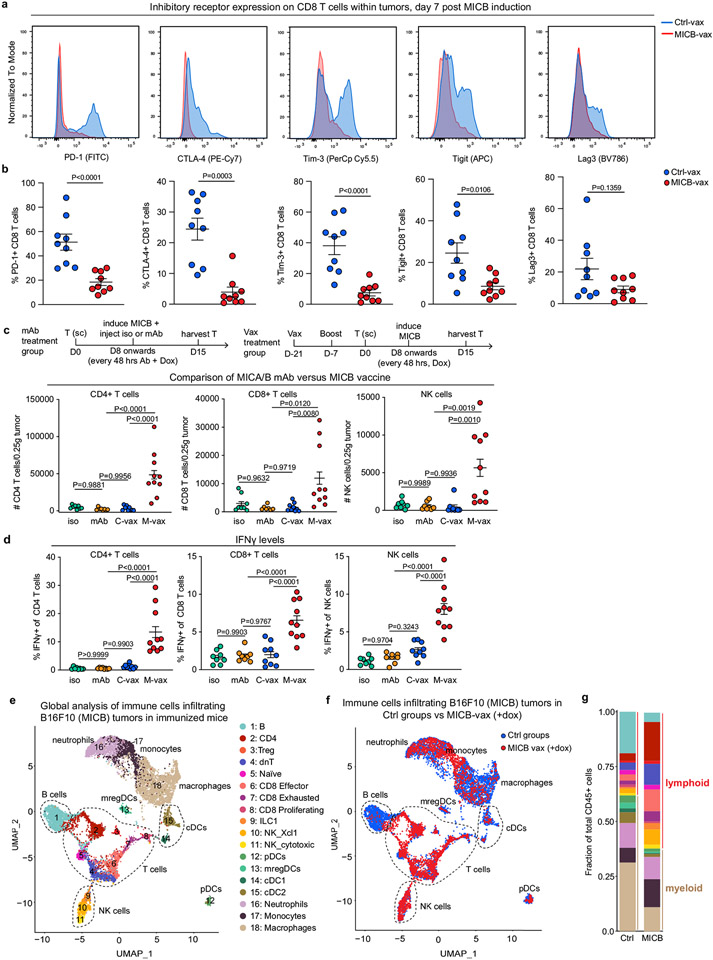
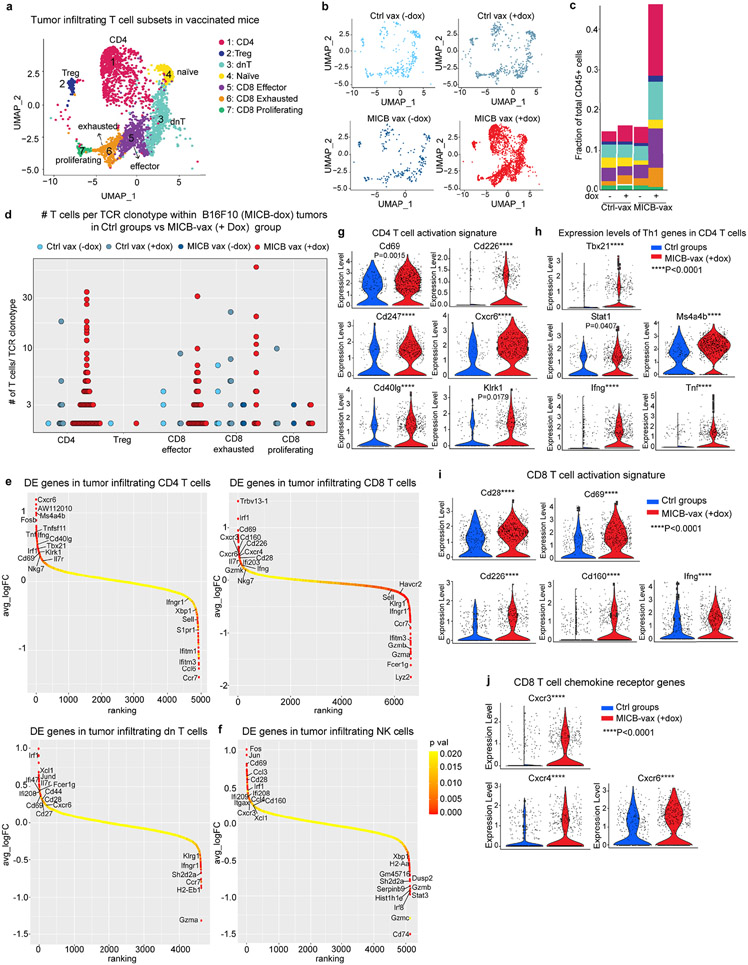
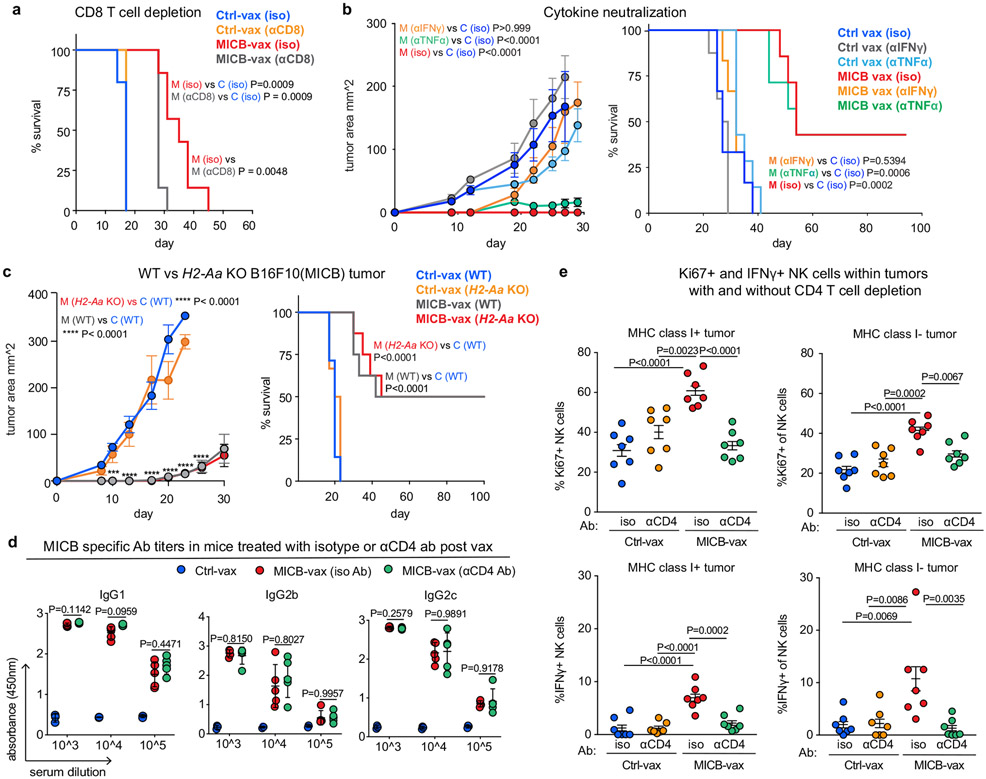
Most cancer vaccines target peptide antigens, necessitating personalization owing to the vast inter-individual diversity in major histocompatibility complex (MHC) molecules that present peptides to T cells. Furthermore, tumours frequently escape T cell-mediated immunity through mechanisms that interfere with peptide presentation1. Here we report a cancer vaccine that induces a coordinated attack by diverse T cell and natural killer (NK) cell populations. The vaccine targets the MICA and MICB (MICA/B) stress proteins expressed by many human cancers as a result of DNA damage2. MICA/B serve as ligands for the activating NKG2D receptor on T cells and NK cells, but tumours evade immune recognition by proteolytic MICA/B cleavage3,4. Vaccine-induced antibodies increase the density of MICA/B proteins on the surface of tumour cells by inhibiting proteolytic shedding, enhance presentation of tumour antigens by dendritic cells to T cells and augment the cytotoxic function of NK cells. Notably, this vaccine maintains efficacy against MHC class I-deficient tumours resistant to cytotoxic T cells through the coordinated action of NK cells and CD4+ T cells. The vaccine is also efficacious in a clinically important setting: immunization following surgical removal of primary, highly metastatic tumours inhibits the later outgrowth of metastases. This vaccine design enables protective immunity even against tumours with common escape mutations.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
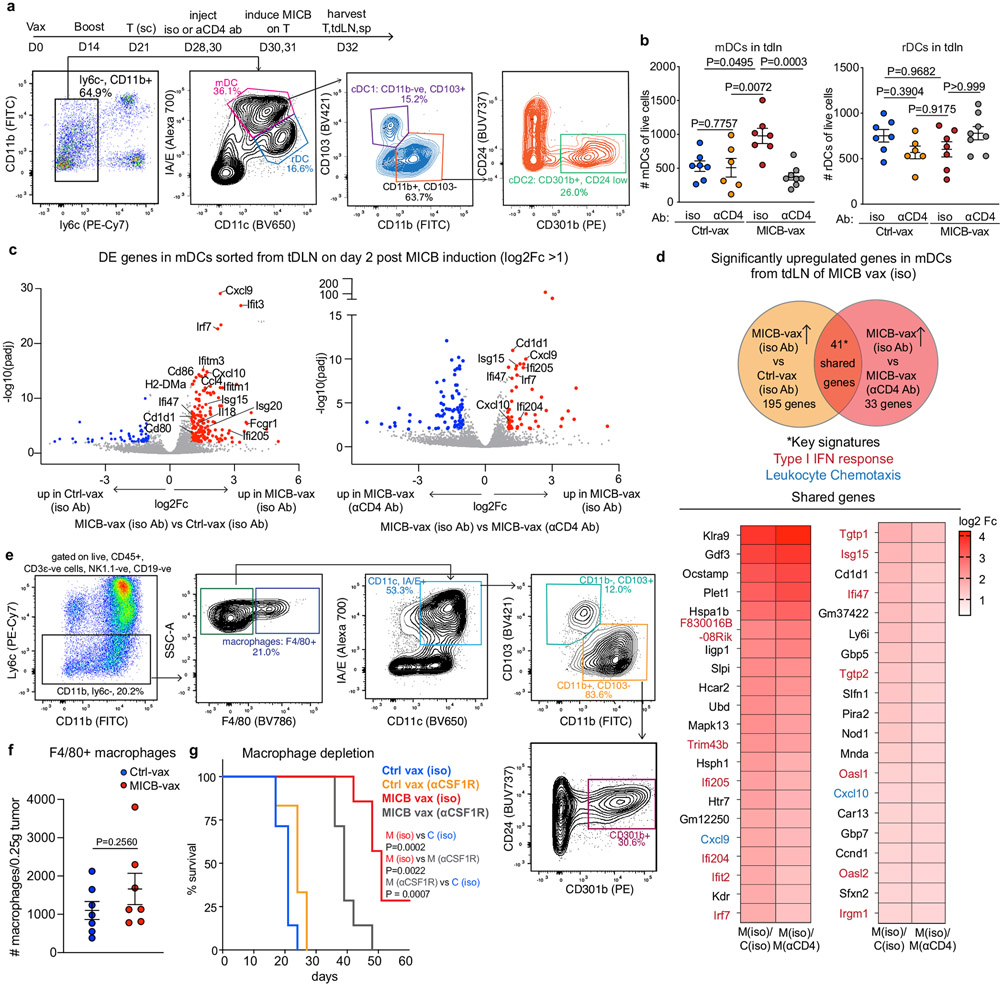
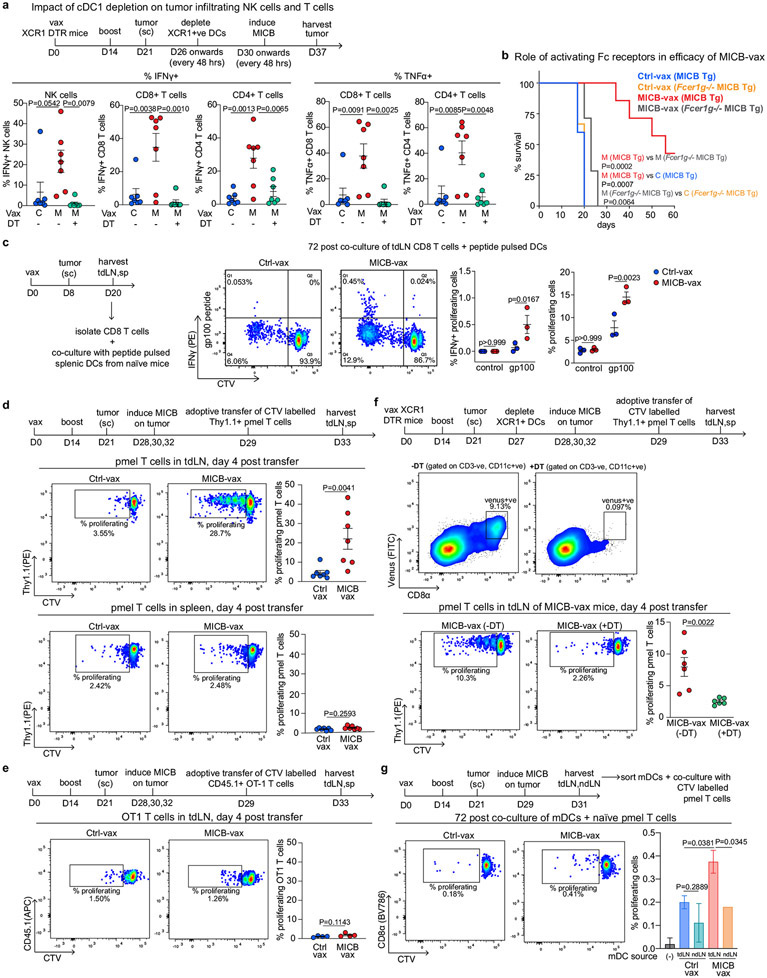
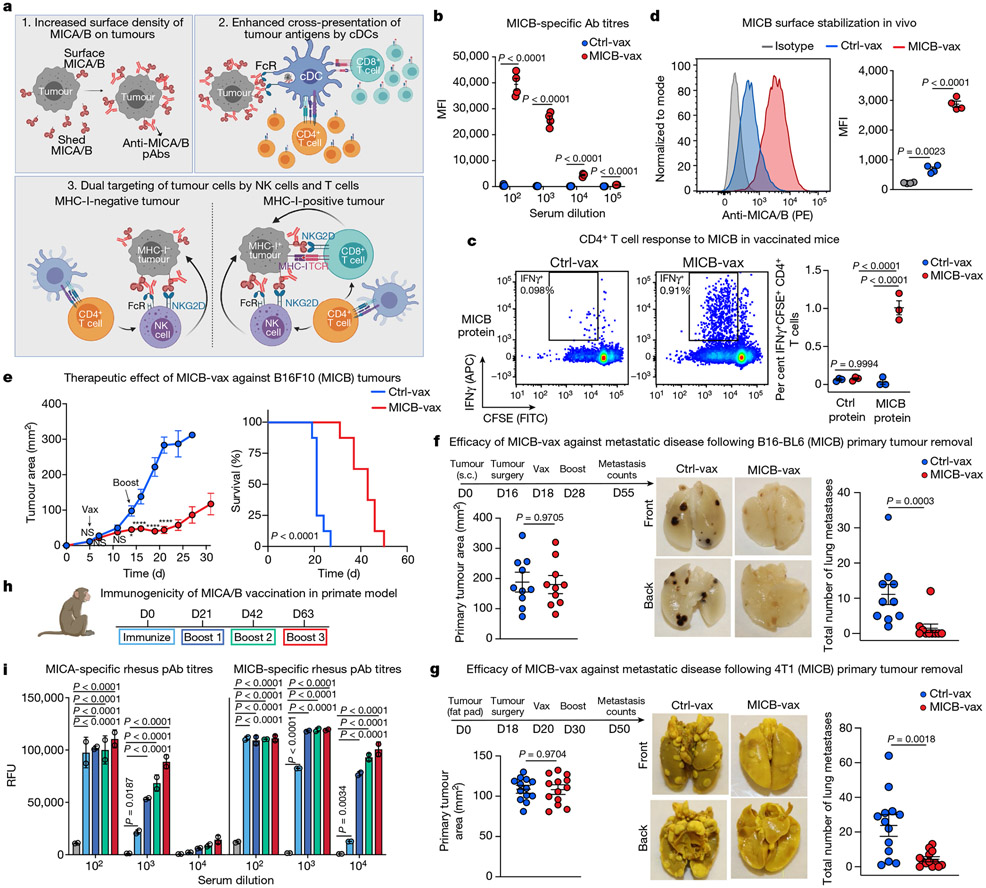
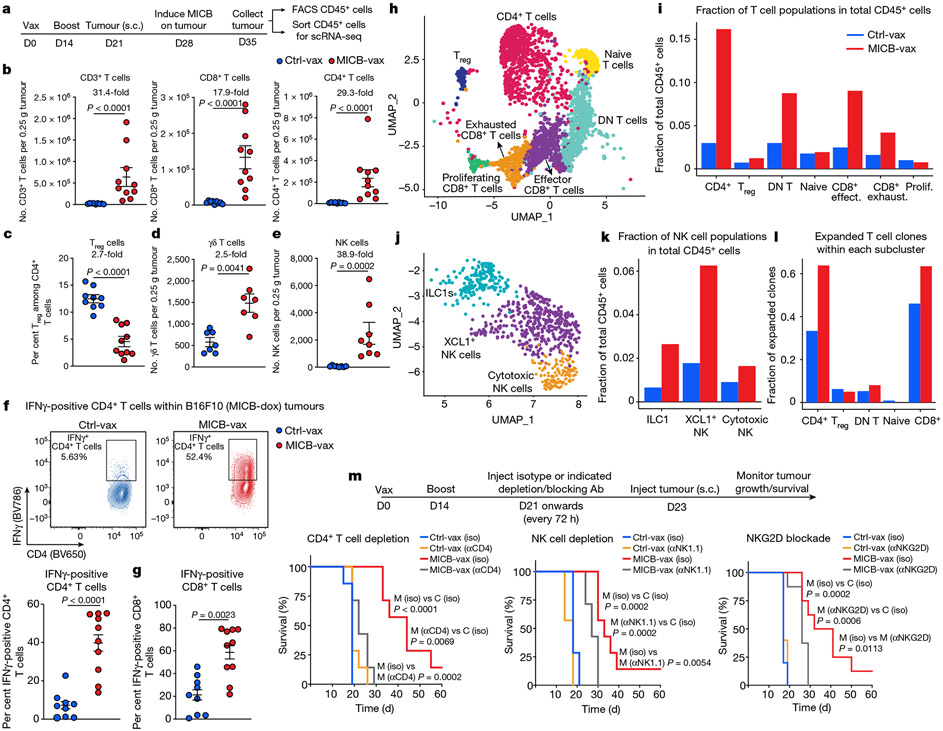

Comment in
-
Vaccine exposes tumours to immune cell attack.Nat Rev Drug Discov. 2022 Jul;21(7):494. doi: 10.1038/d41573-022-00105-2. Nat Rev Drug Discov. 2022. PMID: 35681026 No abstract available.
References
-
- Kaiser BK et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 447, 482–486 (2007). - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
