

Arrhythmias as Presentation of Genetic Cardiomyopathy
- PMID: 35617362
- PMCID: PMC9205615
- DOI: 10.1161/CIRCRESAHA.122.319835
Arrhythmias as Presentation of Genetic Cardiomyopathy
Abstract
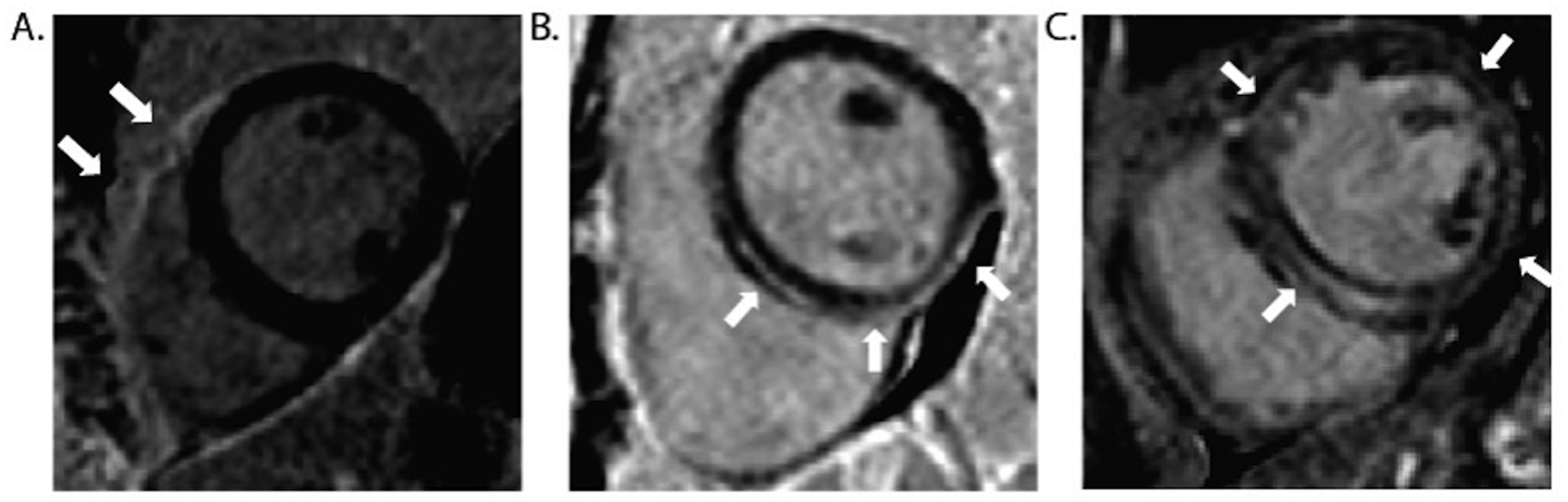
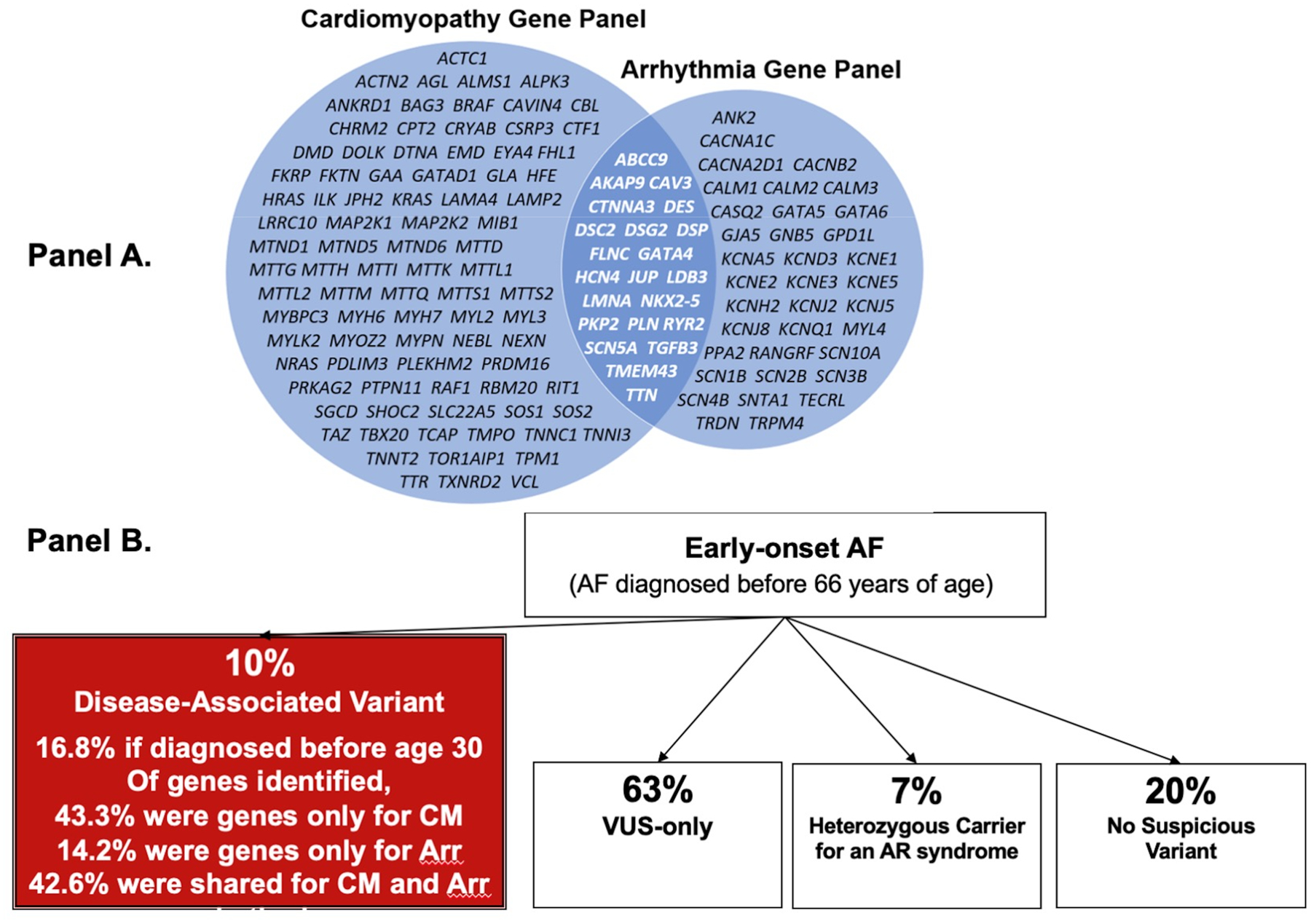
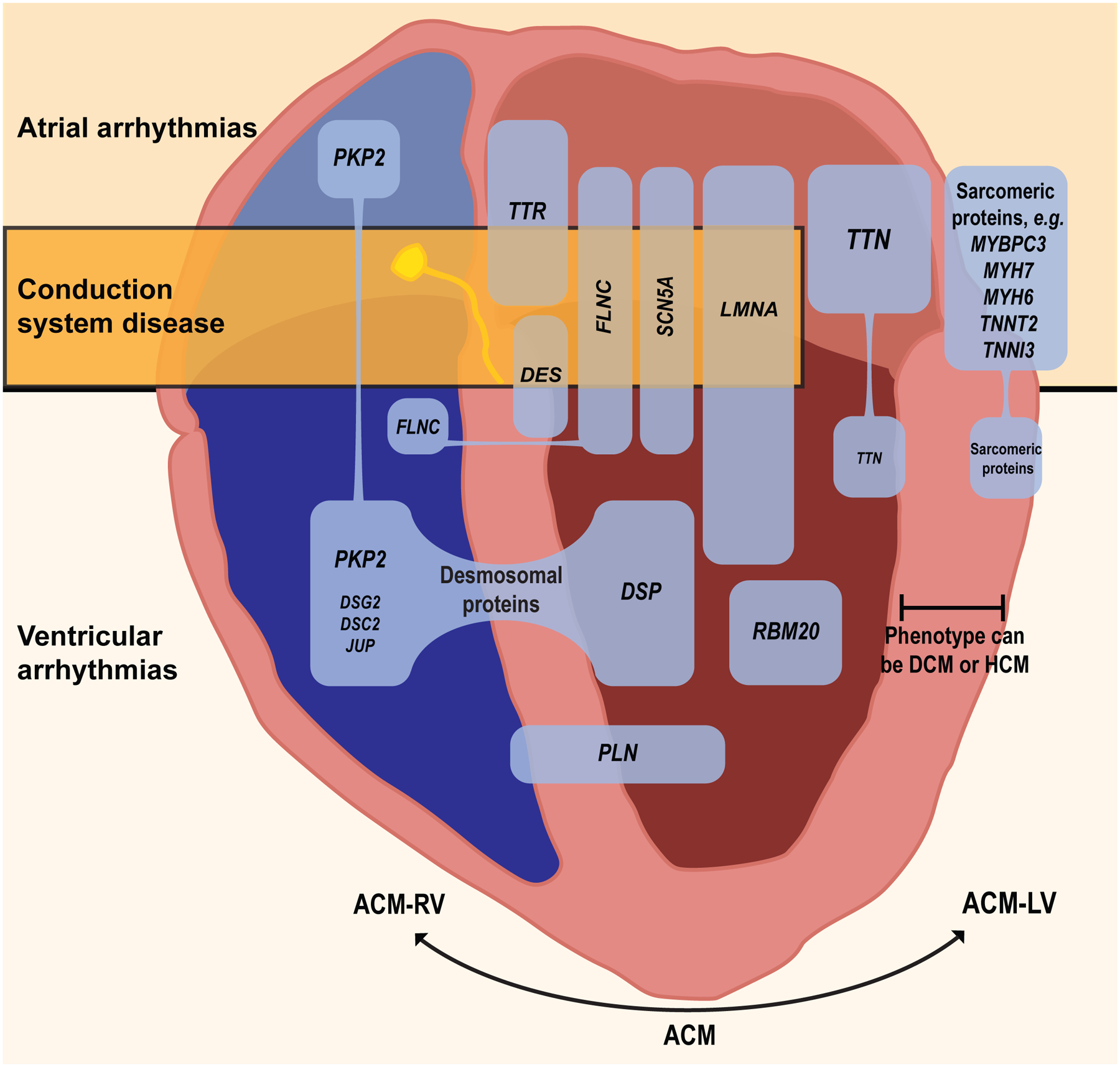
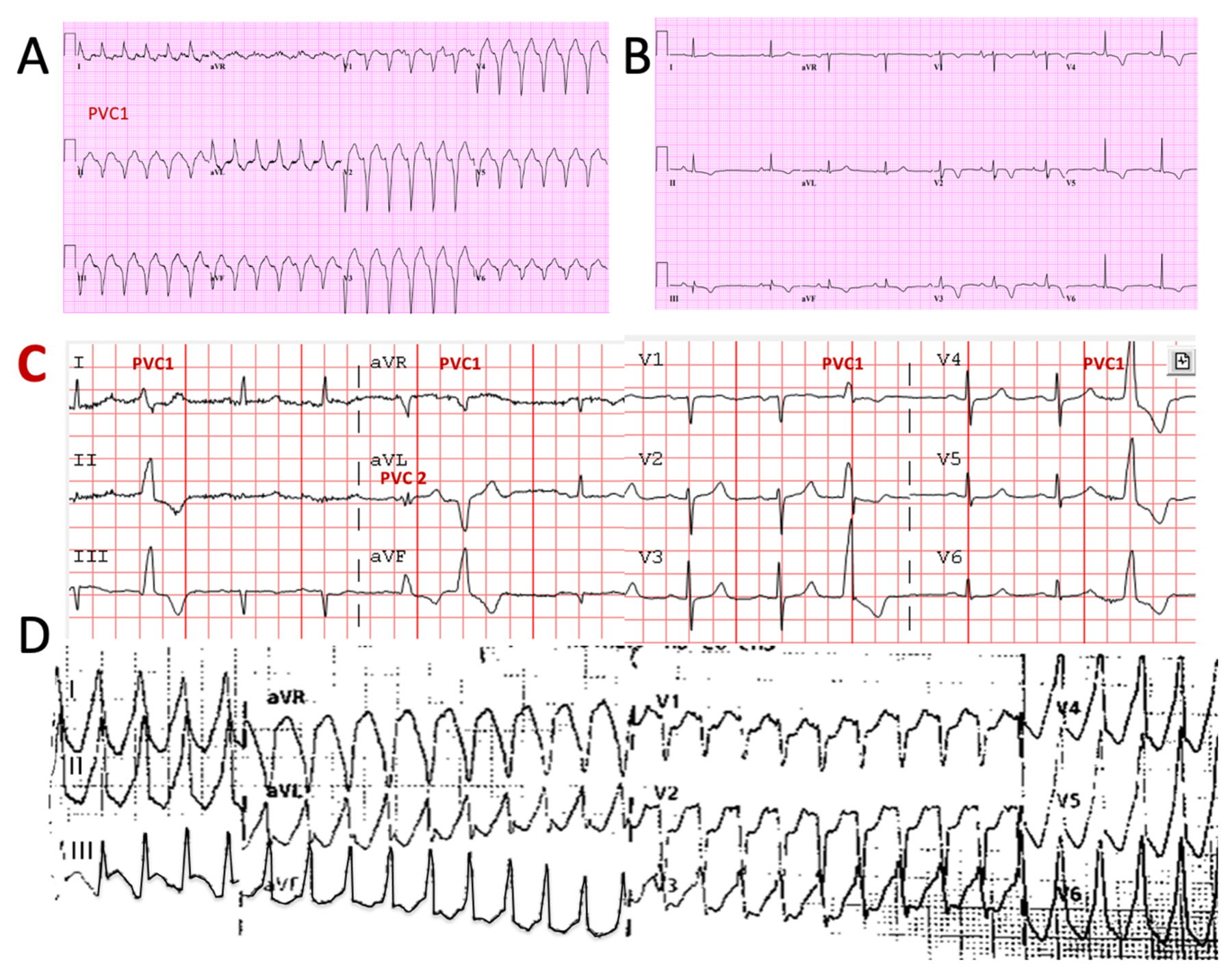
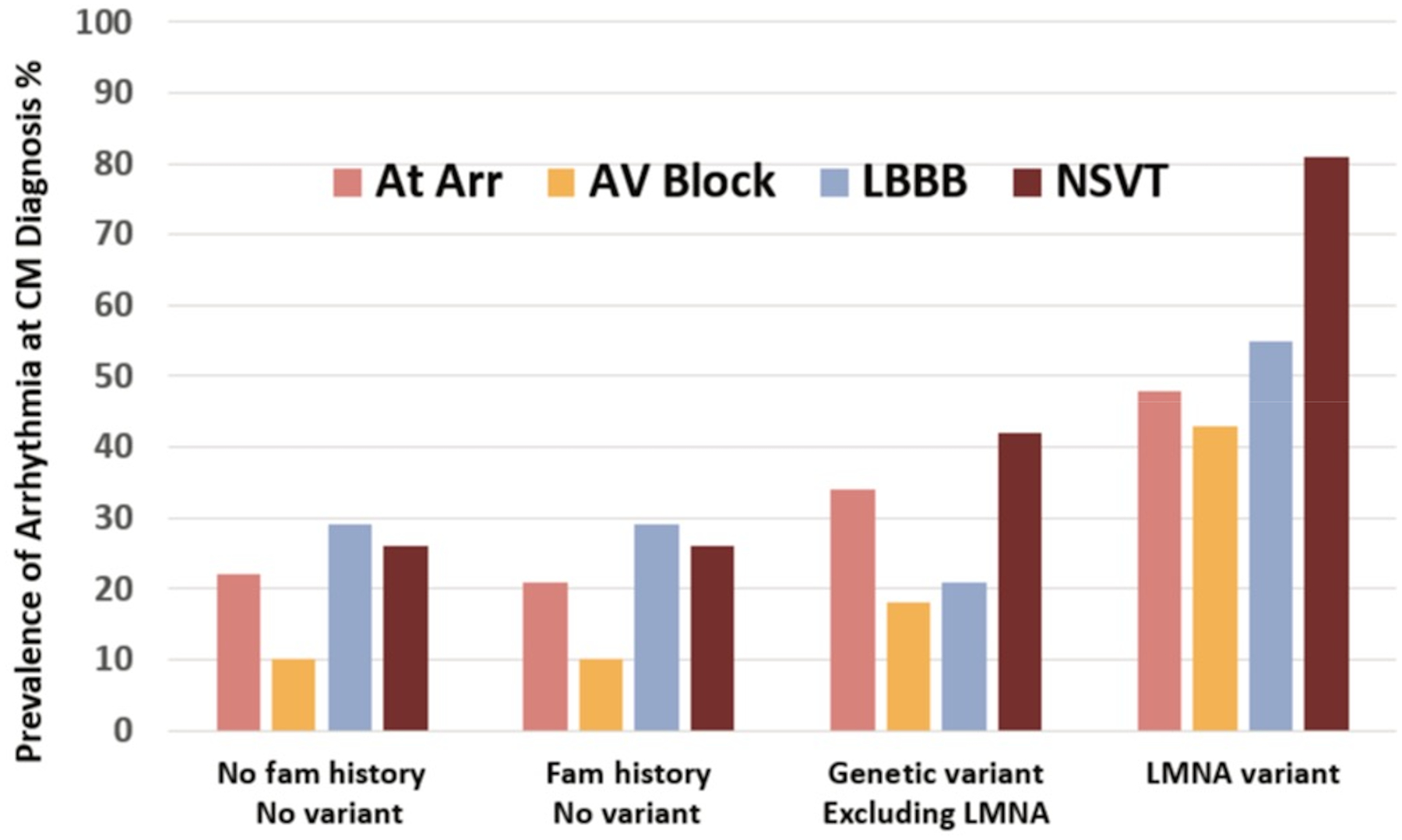
There is increasing evidence regarding the prevalence of genetic cardiomyopathies, for which arrhythmias may be the first presentation. Ventricular and atrial arrhythmias presenting in the absence of known myocardial disease are often labelled as idiopathic, or lone. While ventricular arrhythmias are well-recognized as presentation for arrhythmogenic cardiomyopathy in the right ventricle, the scope of arrhythmogenic cardiomyopathy has broadened to include those with dominant left ventricular involvement, usually with a phenotype of dilated cardiomyopathy. In addition, careful evaluation for genetic cardiomyopathy is also warranted for patients presenting with frequent premature ventricular contractions, conduction system disease, and early onset atrial fibrillation, in which most detected genes are in the cardiomyopathy panels. Sudden death can occur early in the course of these genetic cardiomyopathies, for which risk is not adequately tracked by left ventricular ejection fraction. Only a few of the cardiomyopathy genotypes implicated in early sudden death are recognized in current indications for implantable cardioverter defibrillators which otherwise rely upon a left ventricular ejection fraction ≤0.35 in dilated cardiomyopathy. The genetic diagnoses impact other aspects of clinical management such as exercise prescription and pharmacological therapy of arrhythmias, and new therapies are coming into clinical investigation for specific genetic cardiomyopathies. The expansion of available genetic information and implications raises new challenges for genetic counseling, particularly with the family member who has no evidence of a cardiomyopathy phenotype and may face a potentially negative impact of a genetic diagnosis. Discussions of risk for both probands and relatives need to be tailored to their numeric literacy during shared decision-making. For patients presenting with arrhythmias or cardiomyopathy, extension of genetic testing and its implications will enable cascade screening, intervention to change the trajectory for specific genotype-phenotype profiles, and enable further development and evaluation of emerging targeted therapies.
Keywords: atrial fibrillation; dilated cardiomyopathy; heart failure; phenotype; premature ventricular complexes.
Figures
References
-
- Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–1429. doi: 10.1093/eurheartj/ehz669 - DOI - PMC - PubMed
-
- Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 - DOI - PubMed
-
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–239. doi: 10.1016/j.jacc.2013.05.019 - DOI - PubMed
-
- Cipriani A, Bauce B, De Lazzari M, Rigato I, Bariani R, Meneghin S, Pilichou K, Motta R, Aliberti C, Thiene G, et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. Journal of the American Heart Association. 2020;9. doi: 10.1161/jaha.119.014628 - DOI - PMC - PubMed
-
- Bui QM, Hong KN, Kraushaar M, Ma GS, Brambatti M, Kahn AM, Battiha CE, Boynton K, Storm G, Mestroni L, et al. Myocardial Strain and Association With Clinical Outcomes in Danon Disease: A Model for Monitoring Progression of Genetic Cardiomyopathies. Journal of the American Heart Association. 2021;10. doi: 10.1161/jaha.121.022544 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
