

RNase H2, mutated in Aicardi-Goutières syndrome, resolves co-transcriptional R-loops to prevent DNA breaks and inflammation
- PMID: 35618715
- PMCID: PMC9135716
- DOI: 10.1038/s41467-022-30604-0
RNase H2, mutated in Aicardi-Goutières syndrome, resolves co-transcriptional R-loops to prevent DNA breaks and inflammation
Abstract
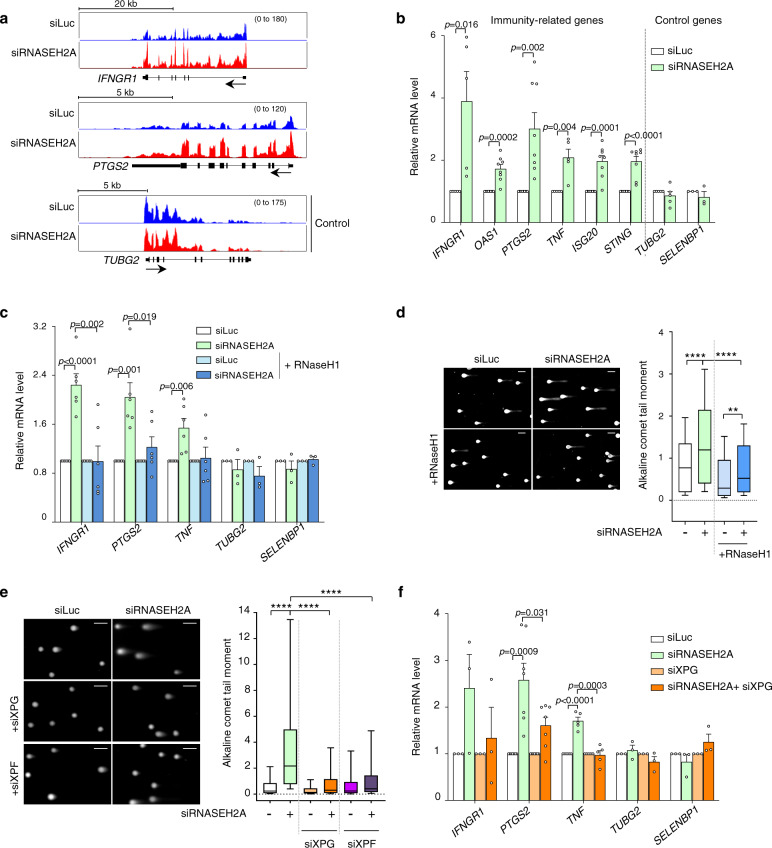
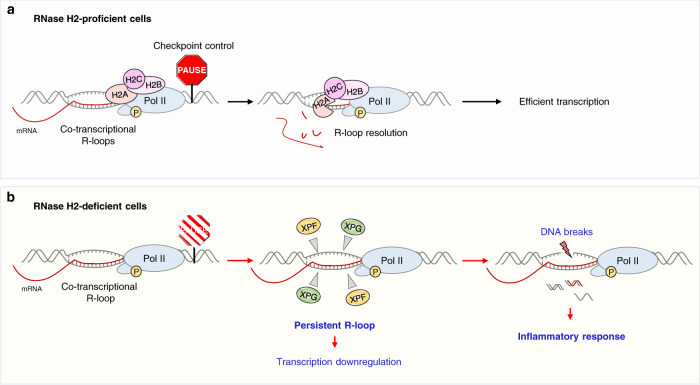
RNase H2 is a specialized enzyme that degrades RNA in RNA/DNA hybrids and deficiency of this enzyme causes a severe neuroinflammatory disease, Aicardi Goutières syndrome (AGS). However, the molecular mechanism underlying AGS is still unclear. Here, we show that RNase H2 is associated with a subset of genes, in a transcription-dependent manner where it interacts with RNA Polymerase II. RNase H2 depletion impairs transcription leading to accumulation of R-loops, structures that comprise RNA/DNA hybrids and a displaced DNA strand, mainly associated with short and intronless genes. Importantly, accumulated R-loops are processed by XPG and XPF endonucleases which leads to DNA damage and activation of the immune response, features associated with AGS. Consequently, we uncover a key role for RNase H2 in the transcription of human genes by maintaining R-loop homeostasis. Our results provide insight into the mechanistic contribution of R-loops to AGS pathogenesis.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
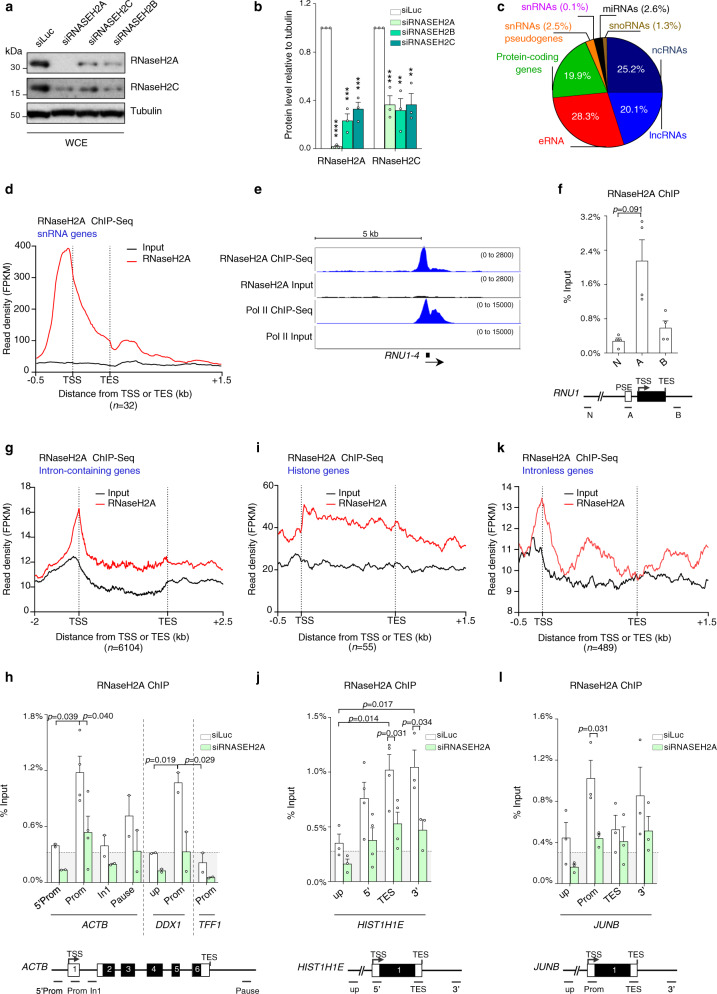
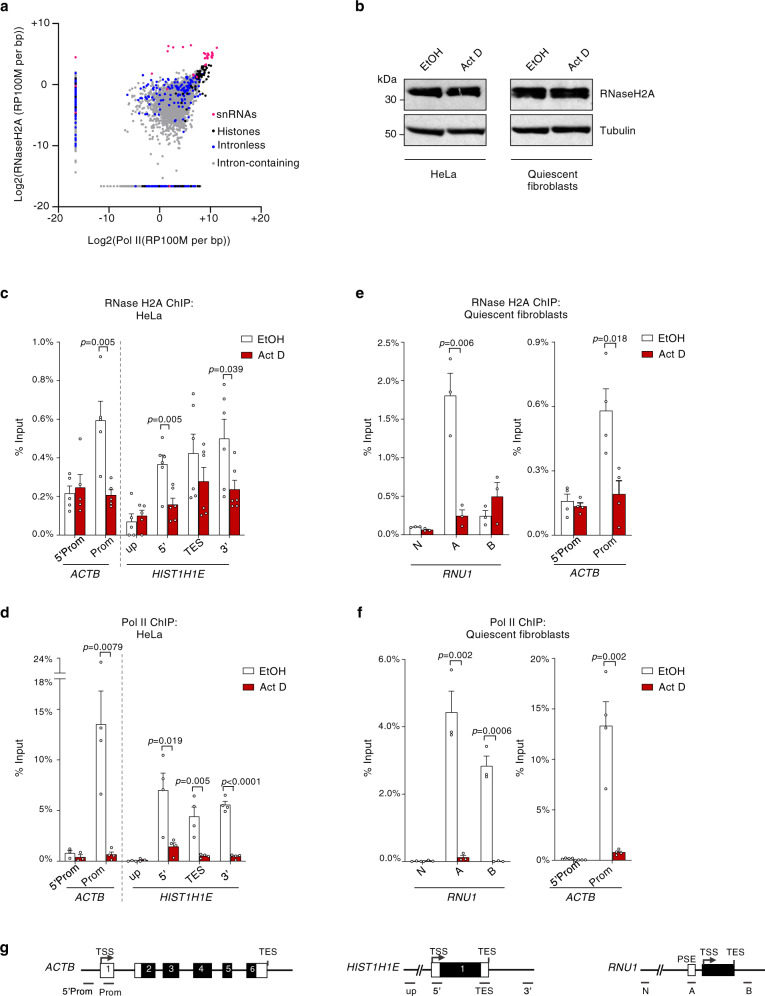
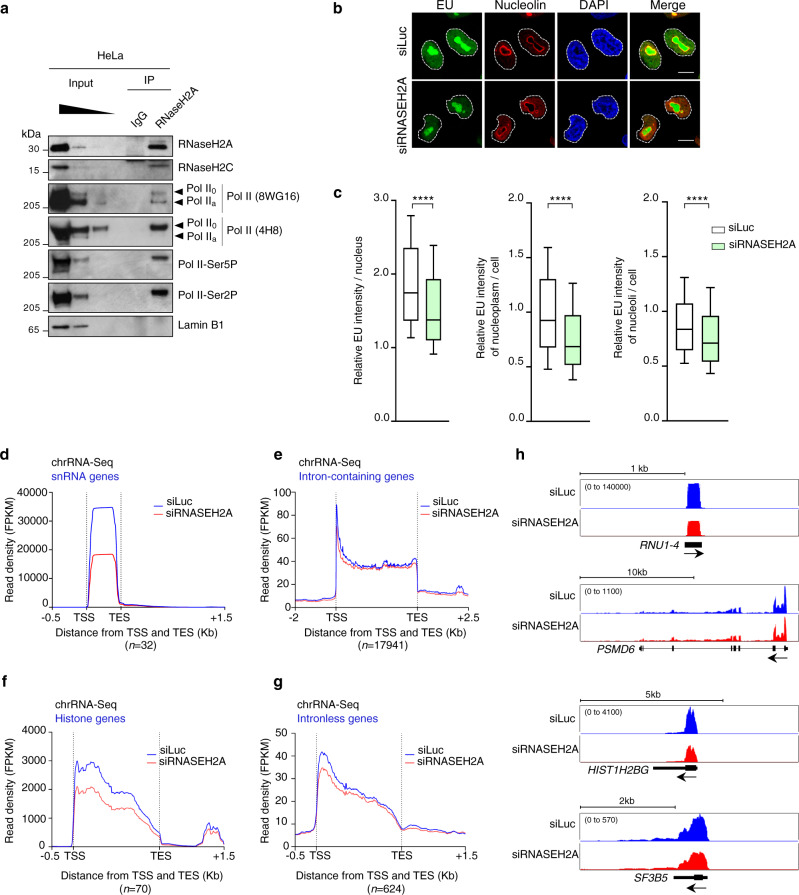
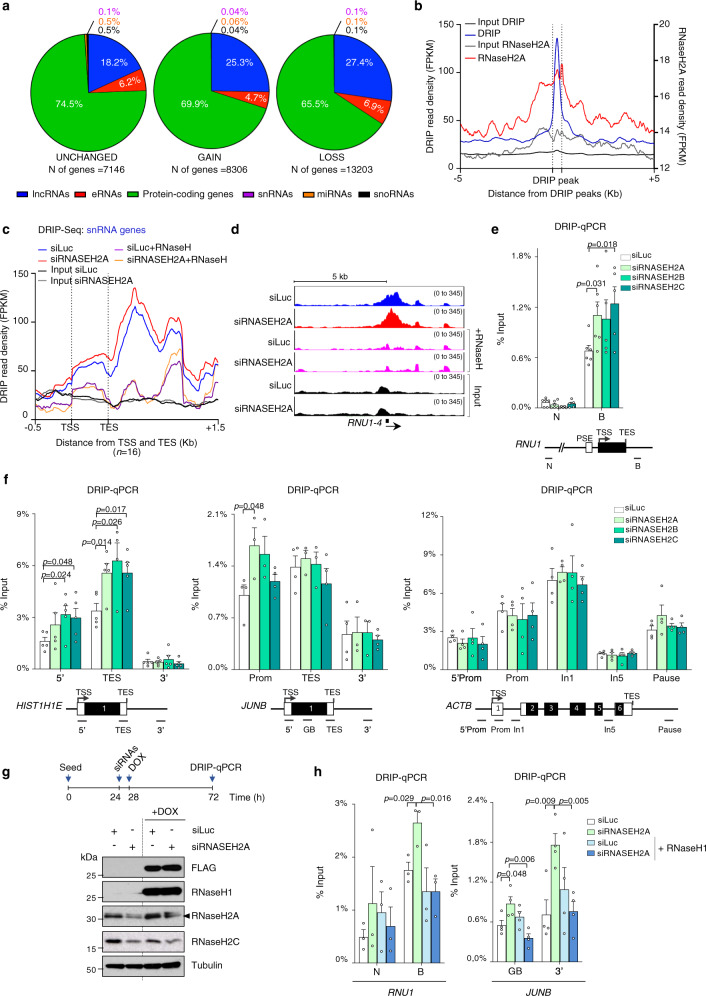
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
