

Genomic analyses of 10,376 individuals in the Westlake BioBank for Chinese (WBBC) pilot project
- PMID: 35618720
- PMCID: PMC9135724
- DOI: 10.1038/s41467-022-30526-x
Genomic analyses of 10,376 individuals in the Westlake BioBank for Chinese (WBBC) pilot project
Abstract
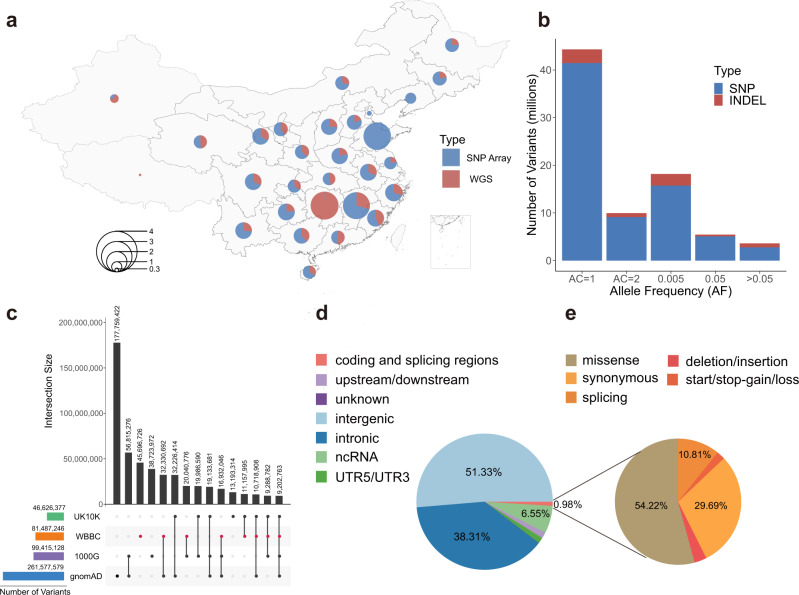
We initiate the Westlake BioBank for Chinese (WBBC) pilot project with 4,535 whole-genome sequencing (WGS) individuals and 5,841 high-density genotyping individuals, and identify 81.5 million SNPs and INDELs, of which 38.5% are absent in dbSNP Build 151. We provide a population-specific reference panel and an online imputation server ( https://wbbc.westlake.edu.cn/ ) which could yield substantial improvement of imputation performance in Chinese population, especially for low-frequency and rare variants. By analyzing the singleton density of the WGS data, we find selection signatures in SNX29, DNAH1 and WDR1 genes, and the derived alleles of the alcohol metabolism genes (ADH1A and ADH1B) emerge around 7,000 years ago and tend to be more common from 4,000 years ago in East Asia. Genetic evidence supports the corresponding geographical boundaries of the Qinling-Huaihe Line and Nanling Mountains, which separate the Han Chinese into subgroups, and we reveal that North Han was more homogeneous than South Han.
© 2022. The Author(s).
Conflict of interest statement
S.-H.Y., W.-W.Z., Y.S., and J.-Q.L. are employees of KingMed Diagnostics Co., Ltd. The other authors have no conflict of interest to declare.
Figures




References
-
- Genetics for all. Nat. Genet. 51, 579 (2019). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
