

Pan-cancer pervasive upregulation of 3' UTR splicing drives tumourigenesis
- PMID: 35618746
- PMCID: PMC9203280
- DOI: 10.1038/s41556-022-00913-z
Pan-cancer pervasive upregulation of 3' UTR splicing drives tumourigenesis
Abstract
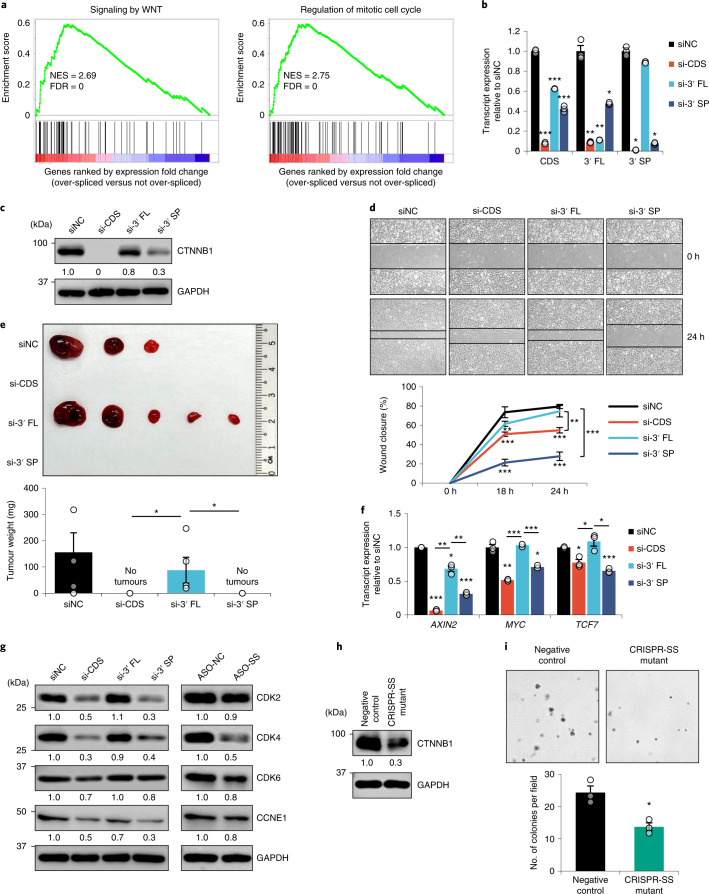
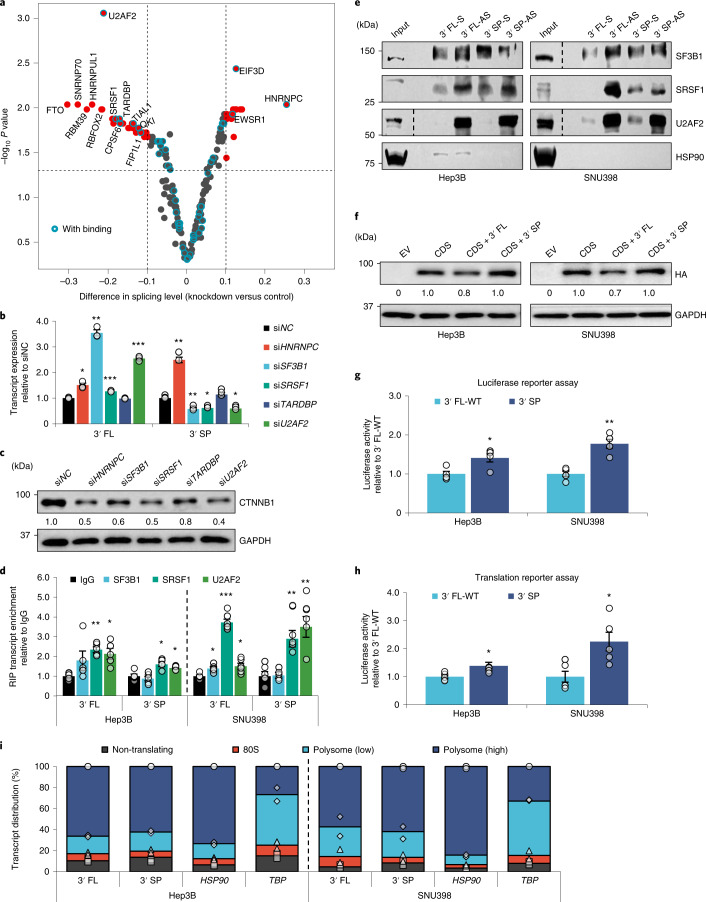
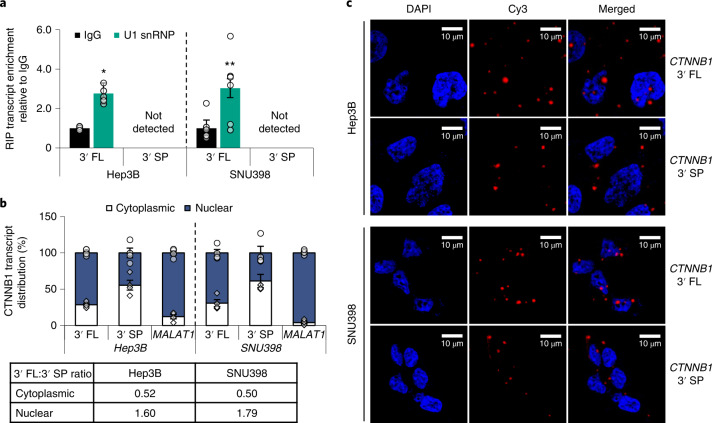
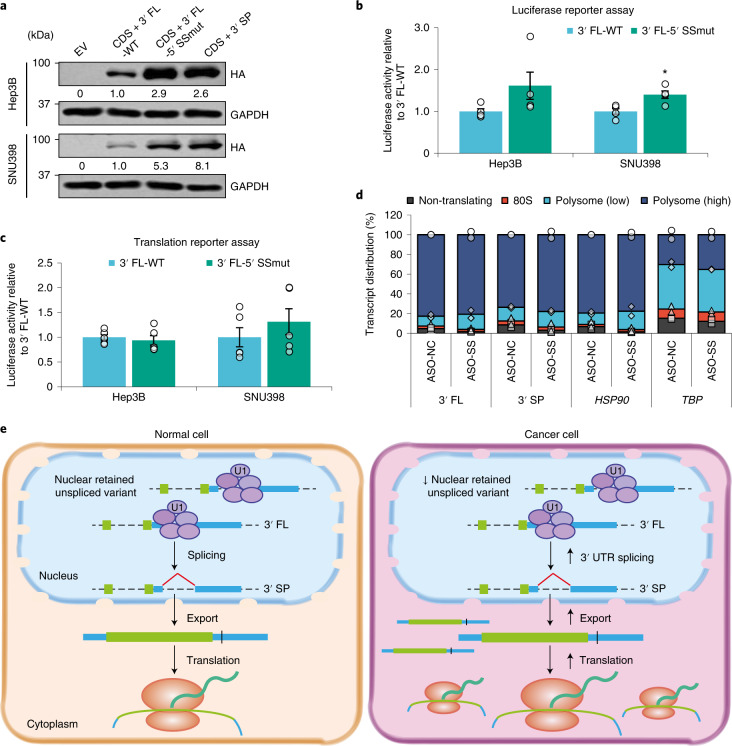
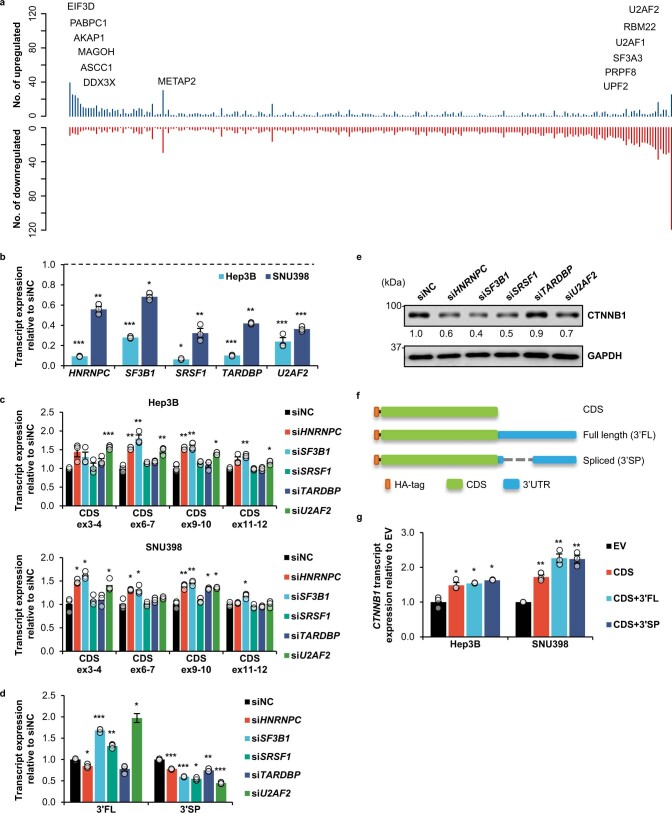
Most mammalian genes generate messenger RNAs with variable untranslated regions (UTRs) that are important post-transcriptional regulators. In cancer, shortening at 3' UTR ends via alternative polyadenylation can activate oncogenes. However, internal 3' UTR splicing remains poorly understood as splicing studies have traditionally focused on protein-coding alterations. Here we systematically map the pan-cancer landscape of 3' UTR splicing and present this in SpUR ( http://www.cbrc.kaust.edu.sa/spur/home/ ). 3' UTR splicing is widespread, upregulated in cancers, correlated with poor prognosis and more prevalent in oncogenes. We show that antisense oligonucleotide-mediated inhibition of 3' UTR splicing efficiently reduces oncogene expression and impedes tumour progression. Notably, CTNNB1 3' UTR splicing is the most consistently dysregulated event across cancers. We validate its upregulation in hepatocellular carcinoma and colon adenocarcinoma, and show that the spliced 3' UTR variant is the predominant contributor to its oncogenic functions. Overall, our study highlights the importance of 3' UTR splicing in cancer and may launch new avenues for RNA-based anti-cancer therapeutics.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
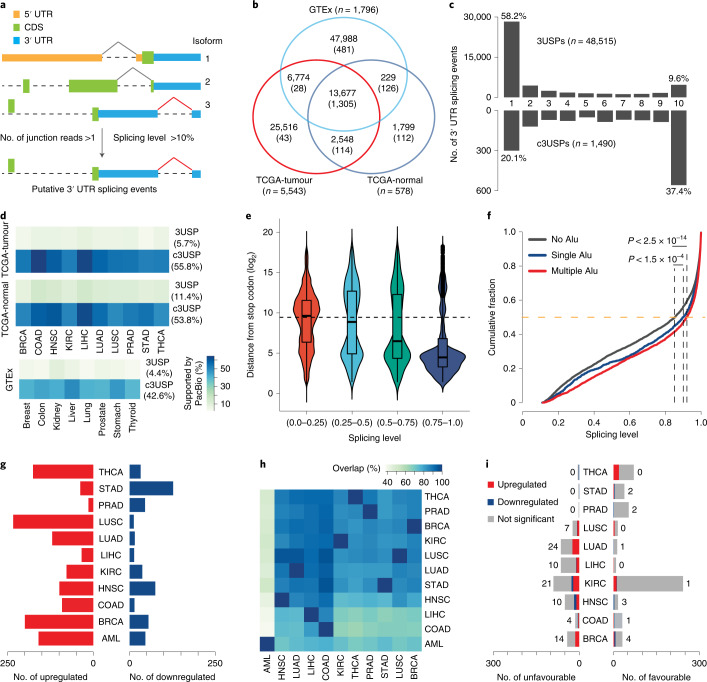
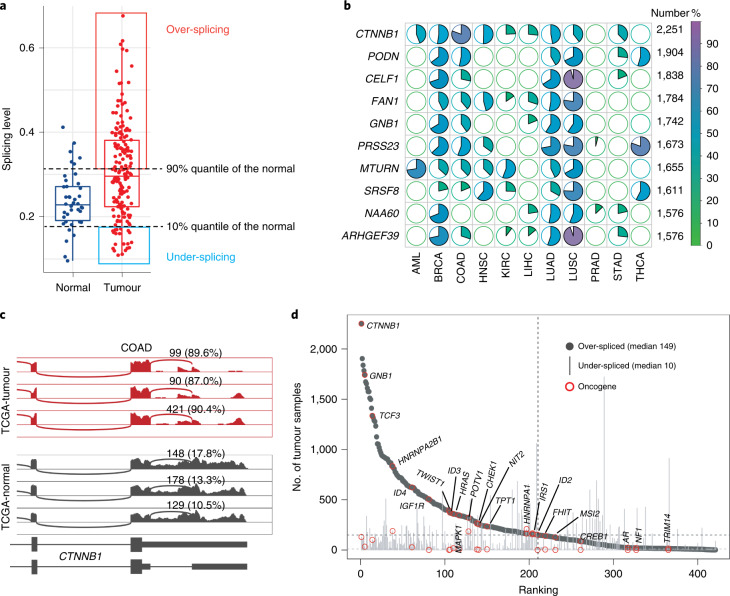
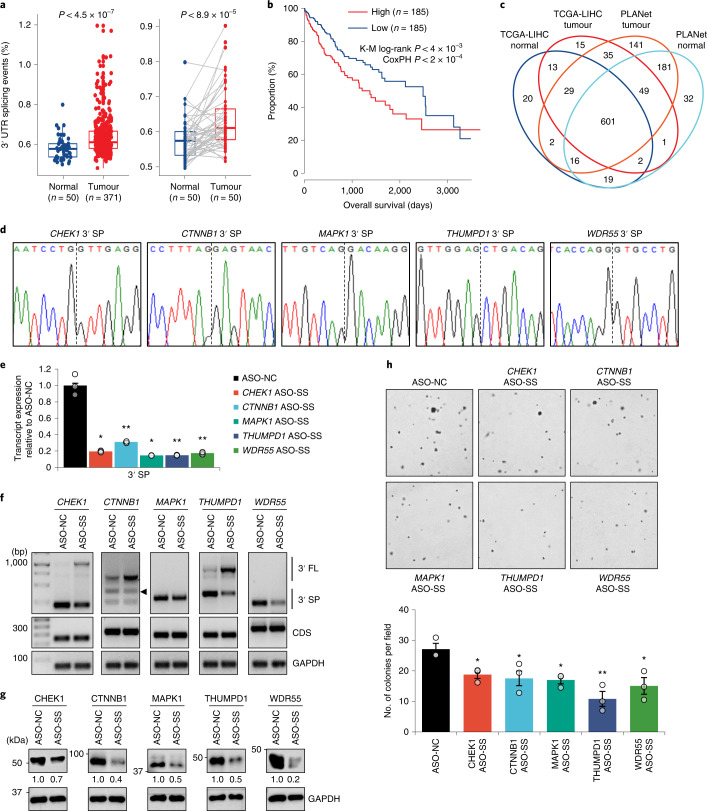
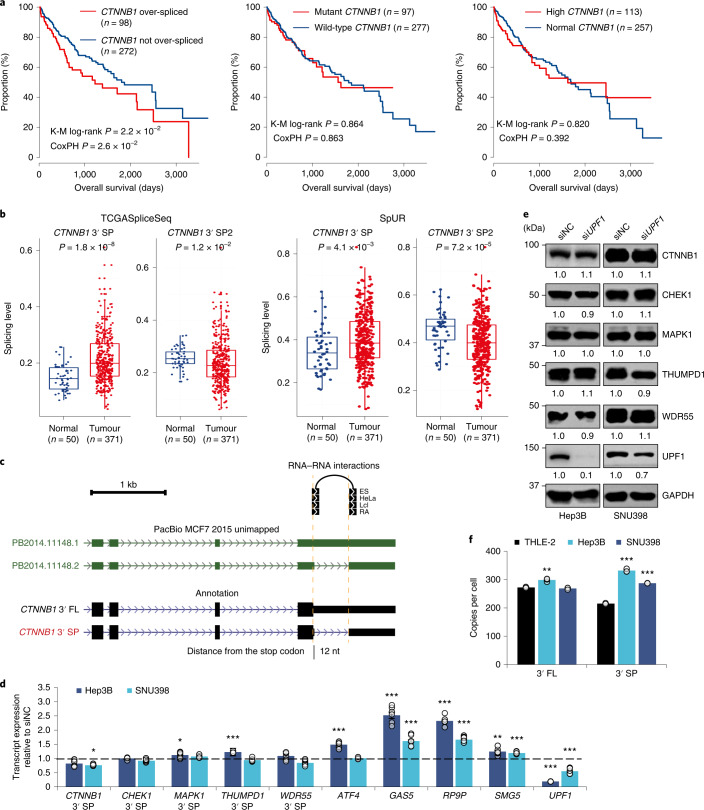









References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
