

Immune disease risk variants regulate gene expression dynamics during CD4+ T cell activation
- PMID: 35618845
- PMCID: PMC9197762
- DOI: 10.1038/s41588-022-01066-3
Immune disease risk variants regulate gene expression dynamics during CD4+ T cell activation
Abstract
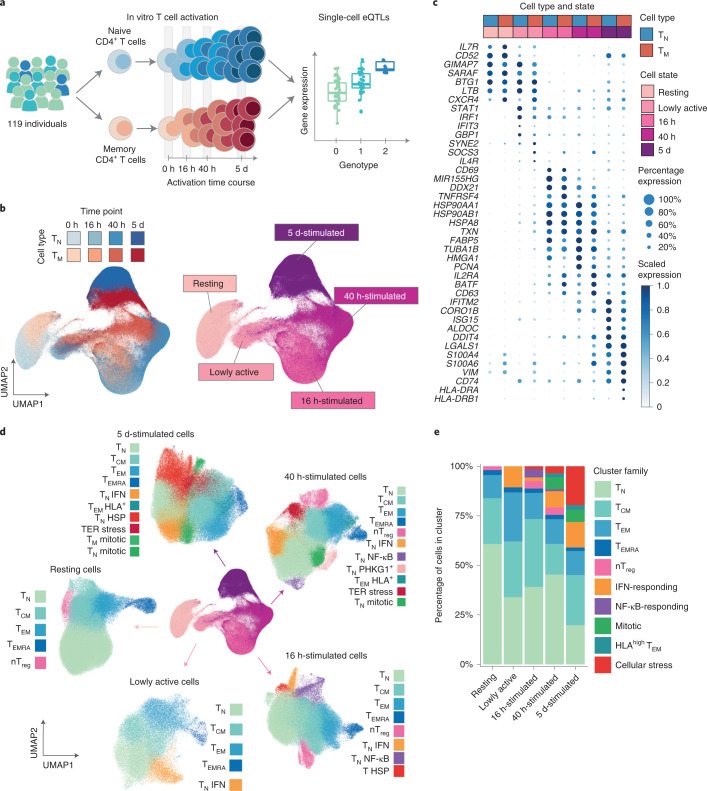
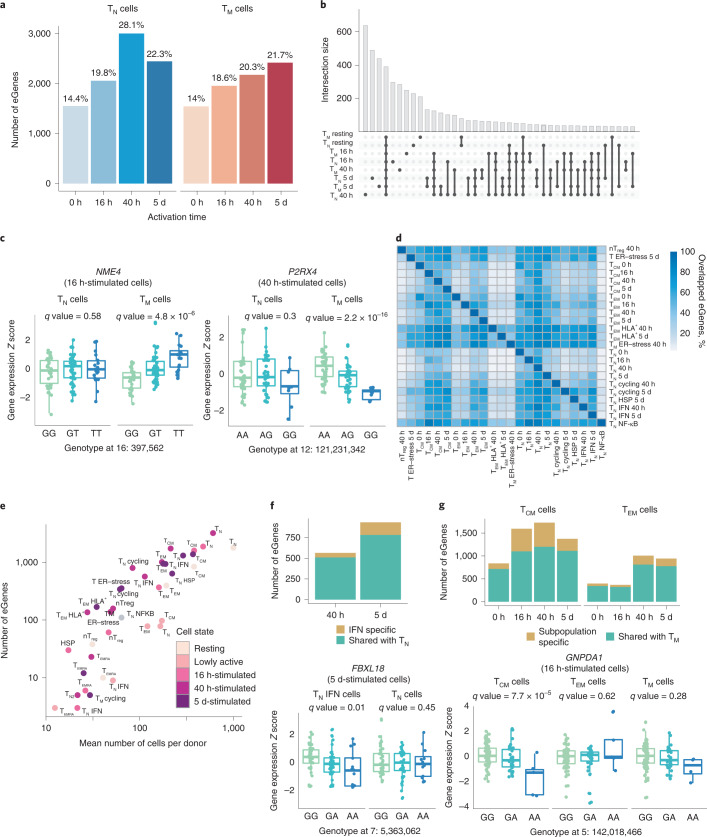
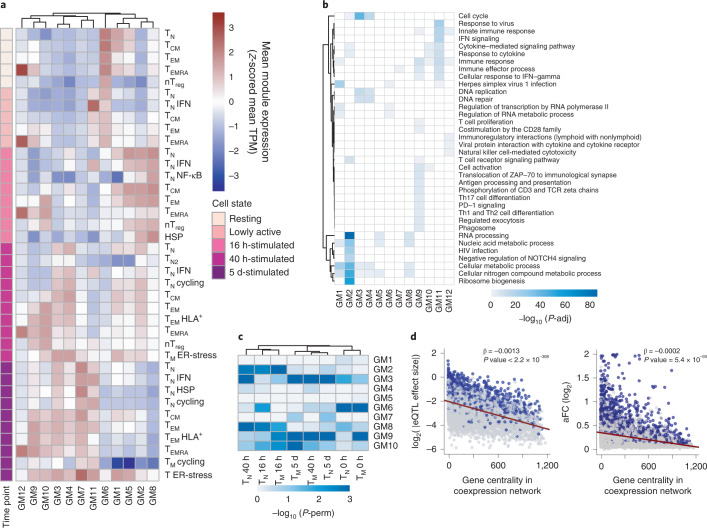
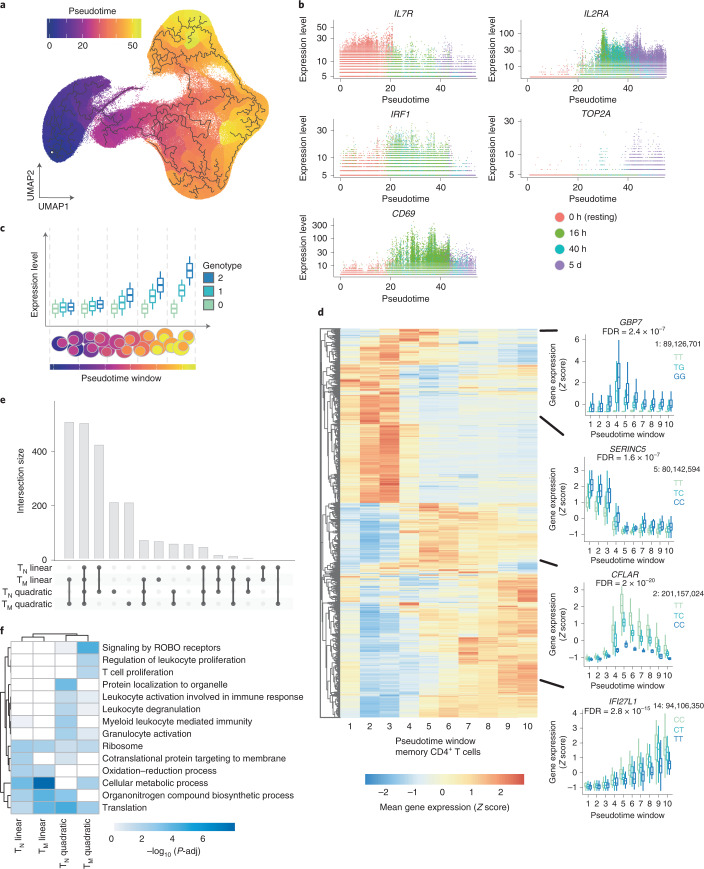
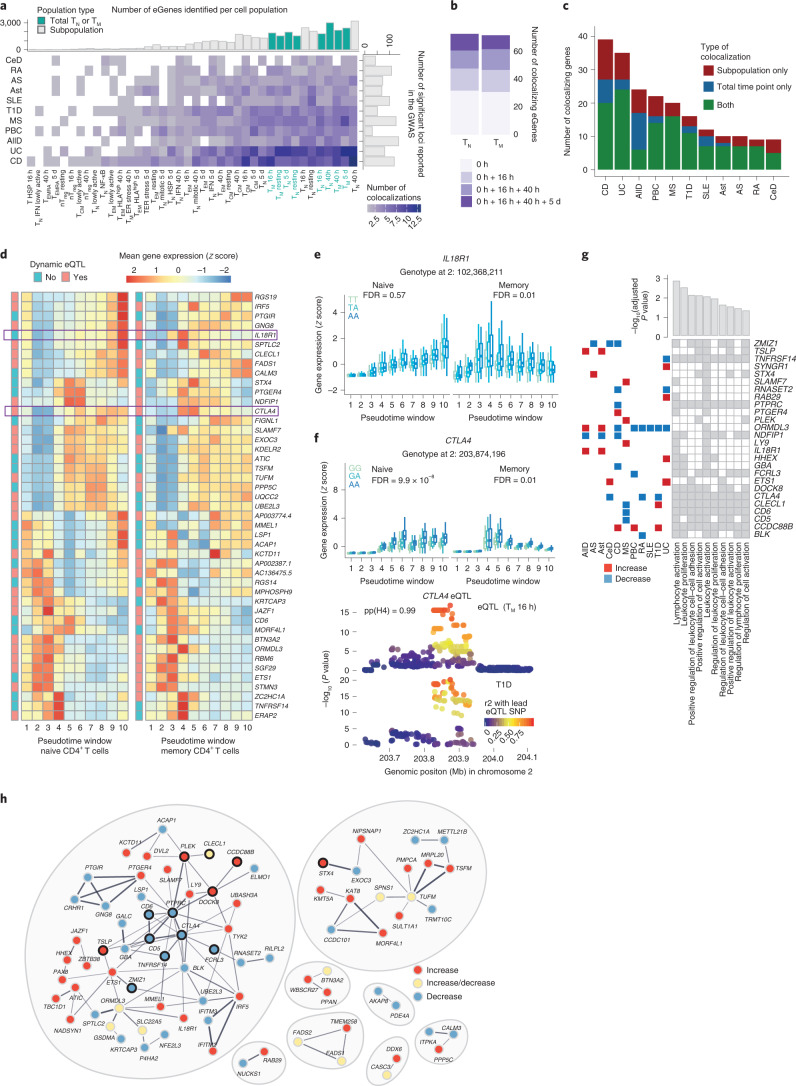
During activation, T cells undergo extensive gene expression changes that shape the properties of cells to exert their effector function. Understanding the regulation of this process could help explain how genetic variants predispose to immune diseases. Here, we mapped genetic effects on gene expression (expression quantitative trait loci (eQTLs)) using single-cell transcriptomics. We profiled 655,349 CD4+ T cells, capturing transcriptional states of unstimulated cells and three time points of cell activation in 119 healthy individuals. This identified 38 cell clusters, including transient clusters that were only present at individual time points of activation. We found 6,407 genes whose expression was correlated with genetic variation, of which 2,265 (35%) were dynamically regulated during activation. Furthermore, 127 genes were regulated by variants associated with immune-mediated diseases, with significant enrichment for dynamic effects. Our results emphasize the importance of studying context-specific gene expression regulation and provide insights into the mechanisms underlying genetic susceptibility to immune-mediated diseases.
© 2022. The Author(s).
Conflict of interest statement
All authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
