

Progress in Amyotrophic Lateral Sclerosis Gene Discovery: Reflecting on Classic Approaches and Leveraging Emerging Technologies
- PMID: 35620141
- PMCID: PMC9128037
- DOI: 10.1212/NXG.0000000000000669
Progress in Amyotrophic Lateral Sclerosis Gene Discovery: Reflecting on Classic Approaches and Leveraging Emerging Technologies
Abstract
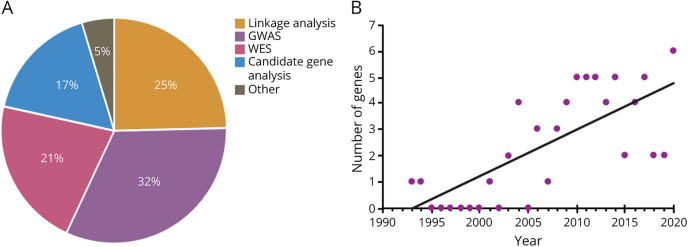
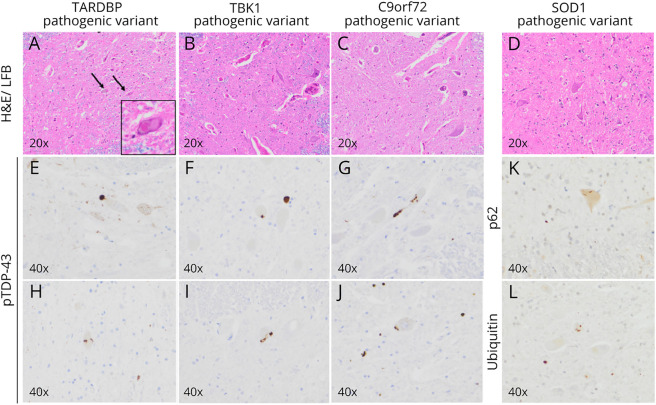
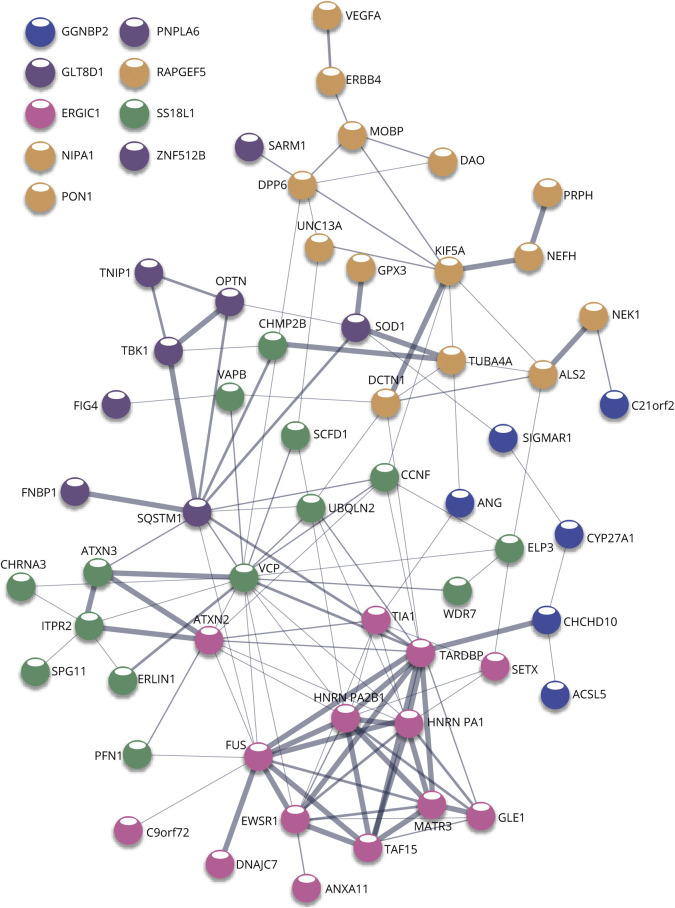
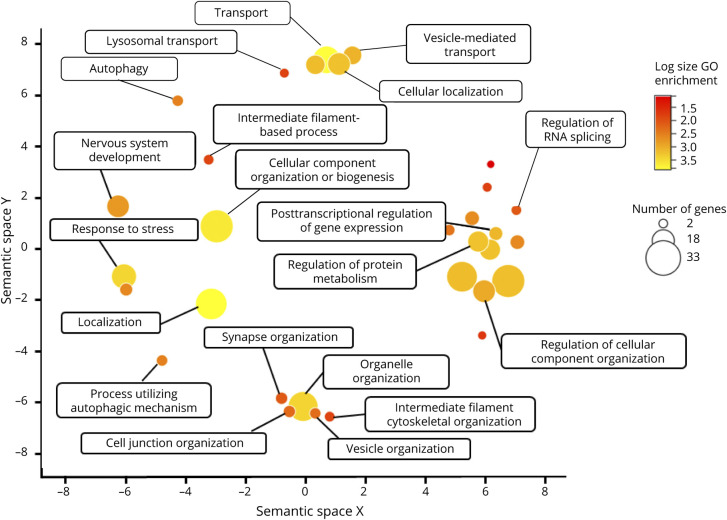
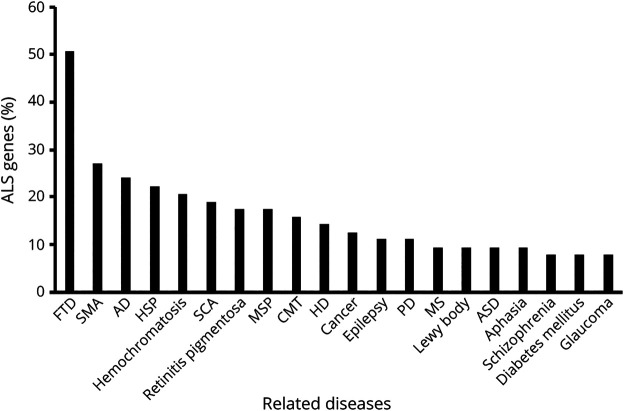
Amyotrophic lateral sclerosis (ALS) is the most prominent motor neuron disease in humans. Its etiology consists of progressive motor neuron degeneration resulting in a rapid decline in motor function starting in the limbs or bulbar muscles and eventually fatally impairing central organs most typically resulting in loss of respiration. Pathogenic variants in 4 main genes, SOD1, TARDBP, FUS, and C9orf72, have been well characterized as causative for more than a decade now. However, these only account for a small fraction of all ALS cases. In this review, we highlight many additional variants that appear to be causative or confer increased risk for ALS, and we reflect on the technologies that have led to these discoveries. Next, we call attention to new challenges and opportunities for ALS and suggest next steps to increase our understanding of ALS genetics. Finally, we conclude with a synopsis of gene therapy paradigms and how increased understanding of ALS genetics can lead us to developing effective treatments. Ultimately, a consolidated update of the field can provide a launching point for researchers and clinicians to improve our search for ALS-related genes, defining pathogenic mechanisms, form diagnostics, and develop therapies.
Copyright © 2022 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
-
- Goldstein LH, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol. 2013;12(4):368–380. - PubMed
-
- Quinn C, Elman L. Amyotrophic lateral sclerosis and other motor neuron diseases. Continuum (Minneap MN). 2020;26(5):1323–1347. - PubMed
-
- Camu W, Khoris J, Moulard B, et al. Genetics of familial ALS and consequences for diagnosis. J Neurol Sci. 1999;165:S21–S26. - PubMed
-
- Sejvar JJ, Holman RC, Bresee JS, Kochanek KD, Schonberger LB. Amyotrophic lateral sclerosis mortality in the United States, 1979-2001. Neuroepidemiology. 2005;25(3):144–152. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous