

Two siblings suffering from Angelman Syndrome with a novel c.1146T>G mutation in UBE3A: A case report
- PMID: 35620312
- PMCID: PMC9112374
- DOI: 10.3892/br.2022.1531
Two siblings suffering from Angelman Syndrome with a novel c.1146T>G mutation in UBE3A: A case report
Abstract
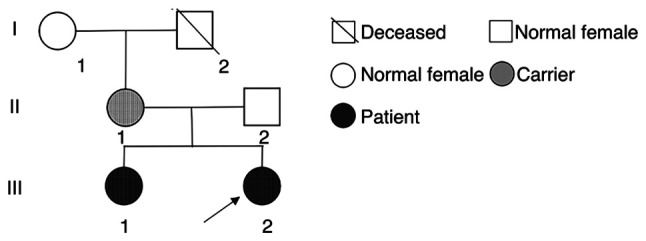
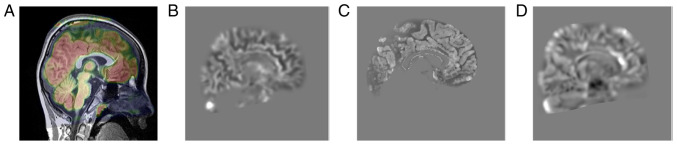
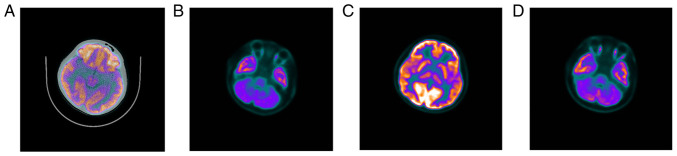
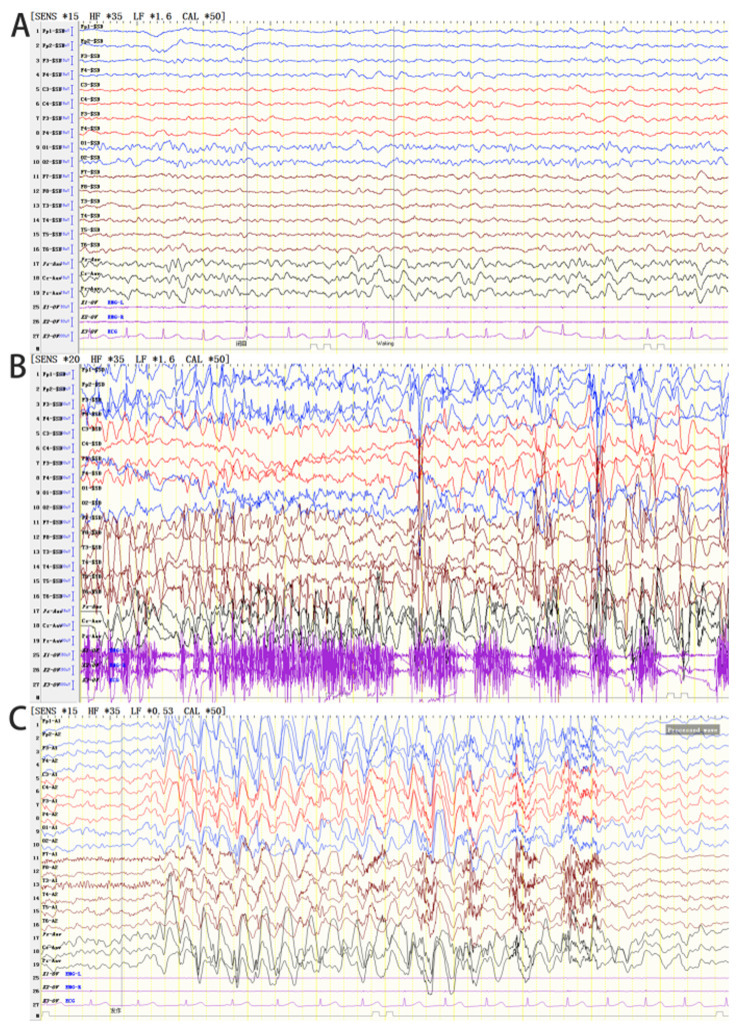
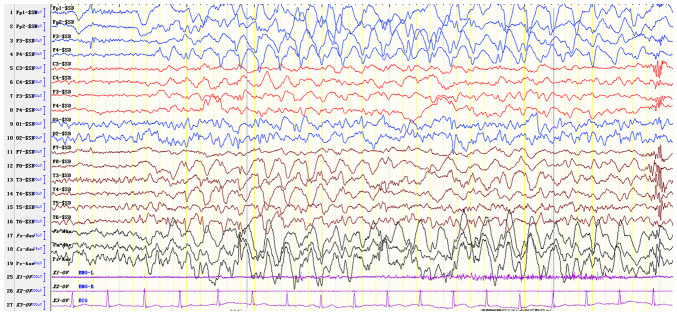
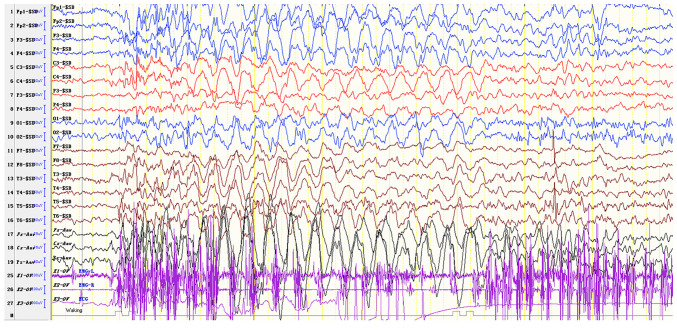
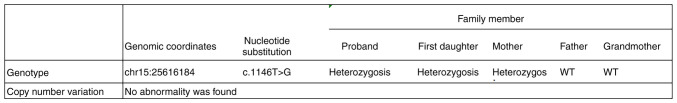
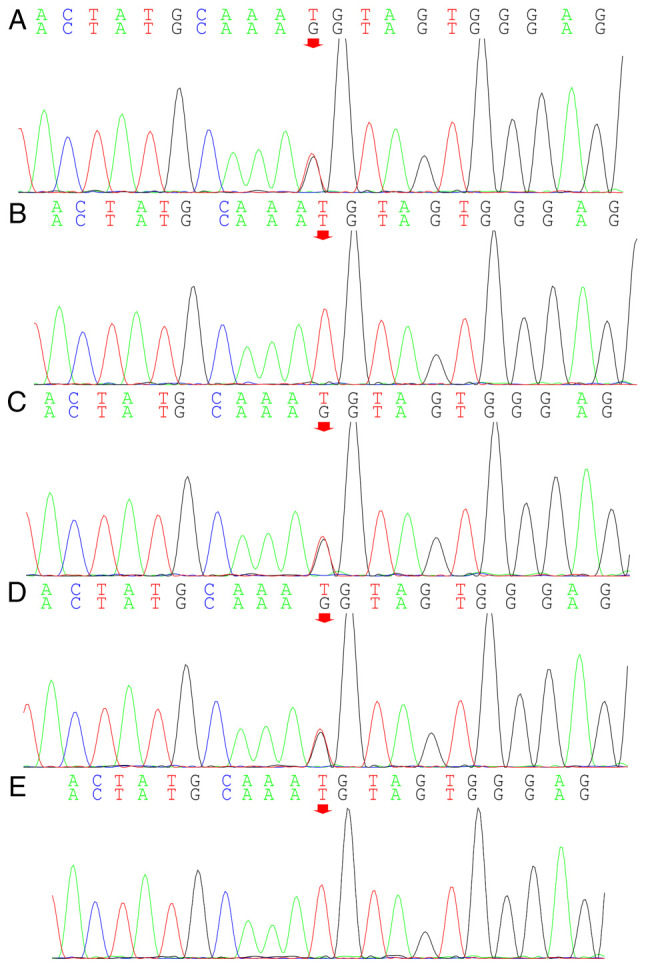
Angelman syndrome (AS) is an autosomal dominant neurodevelopmental genetic disease with maternal imprint, which is associated with the presence of the abnormal chromosome 15q11-q13, and the loss of maternal specific expression of ubiquitin-protein ligase E3A (UBE3A). The expression levels of UBE3A depend on the parental origin and exhibit tissue specificity. In normal brain tissues, the maternal UBE3A gene is actively expressed, whereas the paternal UBE3A gene is not. In total, ~85% of pediatric patients with AS present with epilepsy within their 3rd year of life. This condition is usually difficult to control with medical treatment. An 8-year-old female visited the Affiliated Hospital of Jining Medical University due to frequent epilepsy. Her clinical manifestations included specific facial features, moderate mental retardation and frequent seizures. It was interesting to note that her 15-year-old sister exhibited similar clinical manifestations to those of AS. The results of the electroencephalogram and the imaging examinations were also in line with the characteristics of AS. In order to further clarify the diagnosis, all the suspected genes in her sister and in their parents were sequenced. The multiplex ligation-dependent probe amplification project of the Angel/chubby and copy number variation (CNV) sequencing were assessed concomitantly to identify the pathogenic genes responsible for the development of AS. The latter occurs due to the missense mutation c.1146T>G, which results in asparagine replacement by lysine at position 382 (p.Asn382Lys) in exon 7. This amino acid change affects the normal expression of UBE3A; the mutation is a novel mutation, which, to the best of our knowledge, has not been previously reported. Relevant large fragments of mutations and methylation abnormalities were not found in the associated genes. The data further revealed absence of 25-bp repeat mutations at the shear mutation site of exon 1 of the small nuclear ribonucleoprotein polypeptide N gene in the subjects examined. No suspected CNV was found following analysis.
Keywords: Angelman Syndrome; mutations; pediatric epilepsy; ubiquitin-protein ligase E3A.
Copyright: © Liu et al.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
A novel missense mutation of the ubiquitin protein ligase E3A gene in a patient with Angelman syndrome.Chin Med J (Engl). 2011 Jan;124(1):84-8. Chin Med J (Engl). 2011. PMID: 21362313
-
The spectrum of mutations in UBE3A causing Angelman syndrome.Hum Mol Genet. 1999 Jan;8(1):129-35. doi: 10.1093/hmg/8.1.129. Hum Mol Genet. 1999. PMID: 9887341
-
Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3.PLoS One. 2010 Aug 20;5(8):e12278. doi: 10.1371/journal.pone.0012278. PLoS One. 2010. PMID: 20808828 Free PMC article.
-
Parental imprinting and Angelman syndrome.Adv Neurol. 1999;79:421-9. Adv Neurol. 1999. PMID: 10514831 Review.
-
Angelman syndrome - insights into a rare neurogenetic disorder.Nat Rev Neurol. 2016 Oct;12(10):584-93. doi: 10.1038/nrneurol.2016.133. Epub 2016 Sep 12. Nat Rev Neurol. 2016. PMID: 27615419 Review.
References
-
- Curtis M, Baribeau D, Walker S, Carter M, Costain G, Lamoureux S, Liston E, Marshall CR, Reuter MS, Snell M, et al. A novel intronic variant in UBE3A identified by genome sequencing in a patient with an atypical presentation of Angelman syndrome. Am J Med Genet A. 2020;182:2145–2151. doi: 10.1002/ajmg.a.61740. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials