

doi: 10.1097/HS9.0000000000000724.
eCollection 2022 Jun.
Critical Implications of IVDR for Innovation in Diagnostics: Input From the BioMed Alliance Diagnostics Task Force
Affiliations
- PMID: 35620593
- PMCID: PMC9126521
- DOI: 10.1097/HS9.0000000000000724
Item in Clipboard
Critical Implications of IVDR for Innovation in Diagnostics: Input From the BioMed Alliance Diagnostics Task Force
Hemasphere.
.
No abstract available
Figures
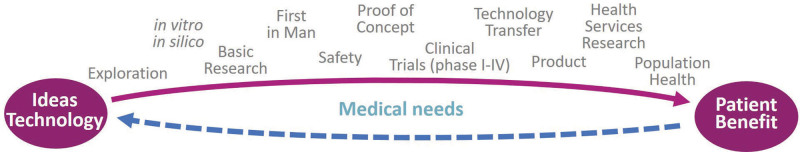
The translational research value chain.
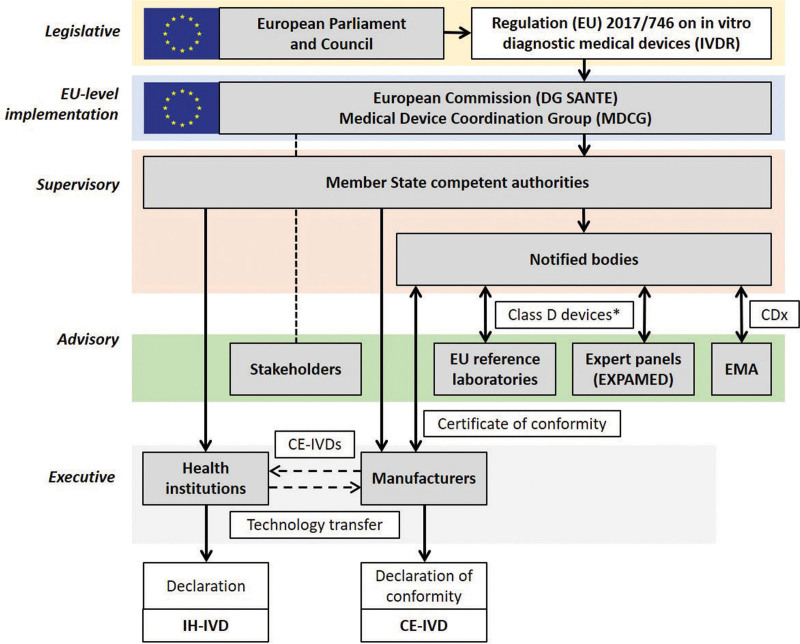
The European IVDR regulatory environment with inclusion of the IH-IVD activities of health institutions. For details on CE-IVDs, see Cobbaert et al. EU level implementation of the IVDR, adopted by the European Parliament and the Council of the EU in 2017, is executed by the European Commission (DG SANTE) and the Medical Device Coordination Group (MDCG), which is chaired by the European Commission and consists of representatives of the Member State competent authorities. Stakeholders such as the BioMed Alliance and EFLM are invited to advise/comment. Supervision of notified bodies, health institutions, and economic operators such as IVD manufacturers is the primary task of the Member State competent authorities. For all IVDs except class A nonsterile devices, manufacturers need to submit their technical documentation to notified bodies for conformity assessment. For class D devices, consultation of EU reference laboratories and/or expert panels (coordinated by the EMA on behalf of the European Commission) can be part of this procedure (*expert panels are consulted when it is the first certification of that type of device and there are no common specifications; EU reference laboratories are involved when a relevant EU reference laboratory is designated; otherwise, it is not mandatory). For CDx, which are typically class C, consultation of the EMA (or a national medicines agency) is included in the conformity assessment procedure by the notified body. After approval, the notified body issues a certificate of conformity, allowing the manufacturer to CE mark the IVD and place it on the market for use in diagnostic patient care. In addition to such CE-IVD tests, health institution diagnostic laboratories can develop and use in-house devices (IH-IVDs), which need to meet a number of conditions and requirements specified in IVDR Article 5.5. Some IH-IVDs might become available for the broad diagnostic community as CE-IVDs after successful technology transfer. CDx = companion diagnostics; CE-IVDs = CE marked in vitro diagnostics; DG SANTE = Directorate-General for Health and Food Safety; EFLM = European Federation of Clinical Chemistry and Laboratory Medicine; EMA = European Medicines Agency; EU = European Union; EXPAMED = expert panel on medical devices and in vitro diagnostic devices; IH-IVDs = in-house in vitro diagnostics; IVD = in vitro diagnostic; IVDR = Regulation (EU) 2017/746 on in vitro diagnostic medical devices; MDCG = Medical Device Coordination Group.
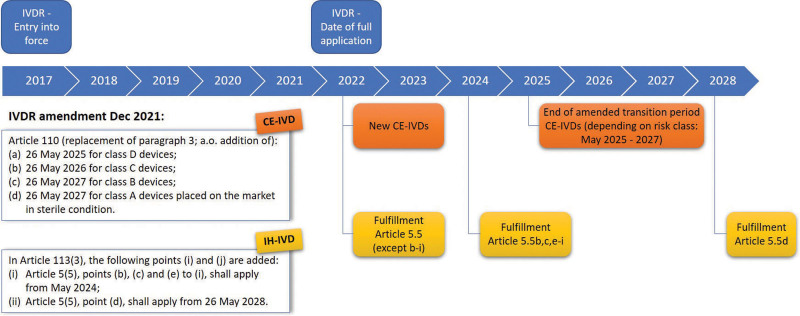
Timelines for revised phased IVDR implementation. The General Safety and Performance Requirements specified in Annex I as well as Article 5.5 (with the exception of 5.5b to i) are not mentioned in the amendment and are, as such, applicable from May 2022. CE-IVDs = CE marked in vitro diagnostics; IH-IVDs = in-house in vitro diagnostics; IVDR = Regulation (EU) 2017/746 on in vitro diagnostic medical devices.
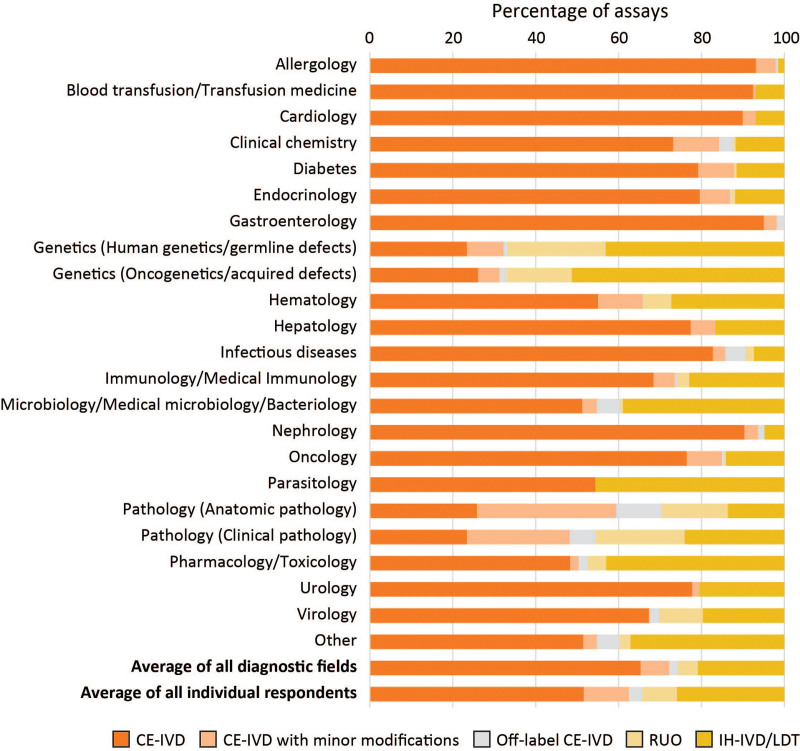
Average percentage of assays from 5 IVD categories used by respondents of the questionnaire (n = 150), per diagnostic field. (1) CE-IVDs used strictly according to the manufacturer’s IFU; (2) CE-IVDs with minor modifications; (3) Off-label CE-IVDs; (4) RUO kits; and (5) In-house devices (IH-IVDs)/LDTs. A detailed explanation of the 5 IVD categories can be can be found in the main text and the report on the BioMed Alliance website. CE-IVDs = CE marked in vitro diagnostics; IFU = instructions for use; IH-IVDs = in-house in vitro diagnostics; IVD = in vitro diagnostic; LDT = laboratory-developed test; RUO = Research Use Only.
References
-
- Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. Official J Eur Commun. 1998;331:1–37.
-
- Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU. Official J Eur Union. 2017;117:176–332.
-
- Lubbers BR, van Dongen JJM. Position of European Consortia in the IVDR Era: Support for In-house Devices (IH-IVDs) and CE Marked IVDs (CE-IVDs). 9th ESLHO Symposium Abstract Book. 2020:33–46. Available at: https://eslho-public.s3.nl-ams.scw.cloud/Lubbers2020_94364d9779.pdf. Accessed March 5, 2022.
-
- Medical Device Coordination Group. MDCG 2022-2 Guidance on General Principles of Clinical Evidence for In Vitro Diagnostic Medical Devices (IVDs). 2022. Available at: https://ec.europa.eu/health/system/files/2022-01/mdcg_2022-2_en.pdf. Accessed March 5, 2022.
LinkOut - more resources
Full Text Sources