

The unfolded protein response to PI*Z alpha-1 antitrypsin in human hepatocellular and murine models
- PMID: 35621045
- PMCID: PMC9426387
- DOI: 10.1002/hep4.1997
The unfolded protein response to PI*Z alpha-1 antitrypsin in human hepatocellular and murine models
Abstract
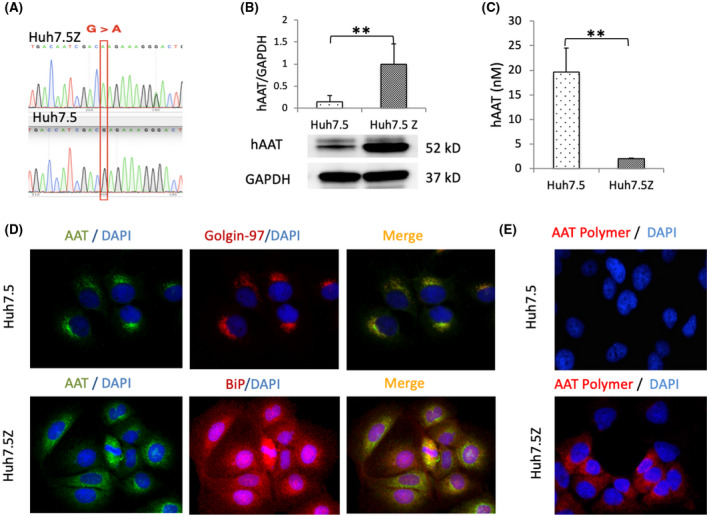
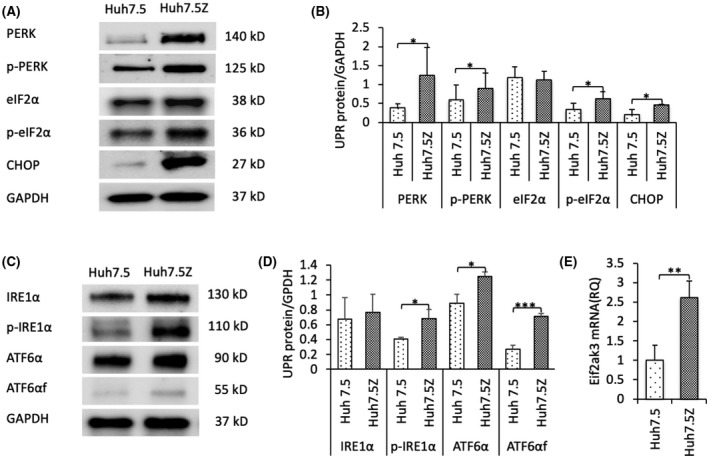
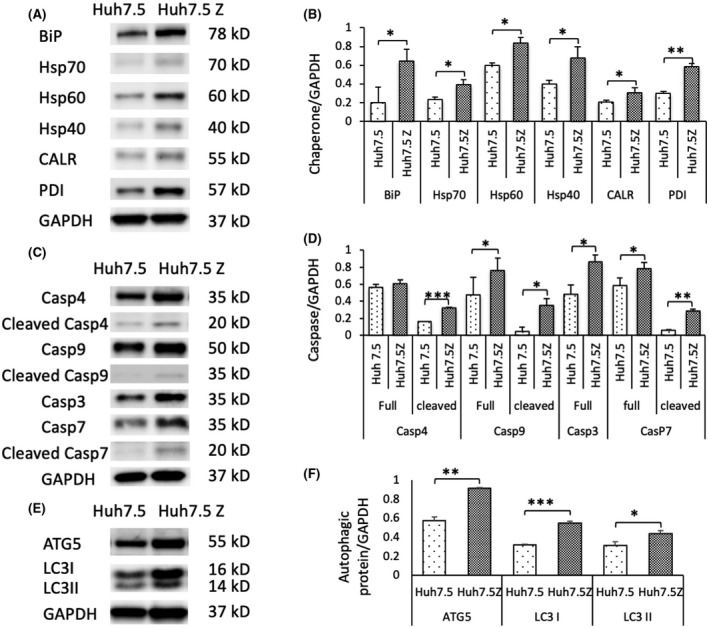
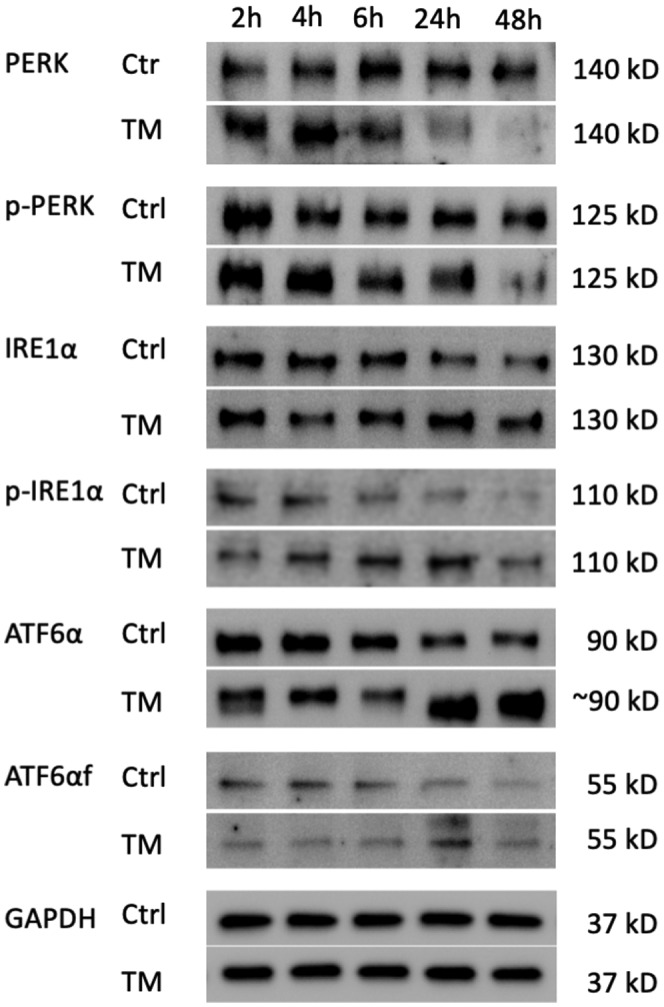
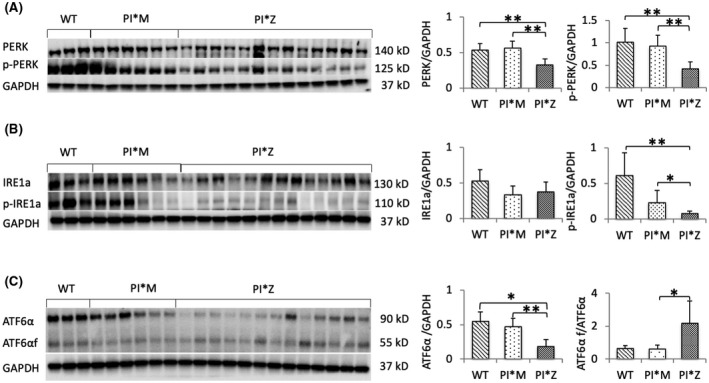
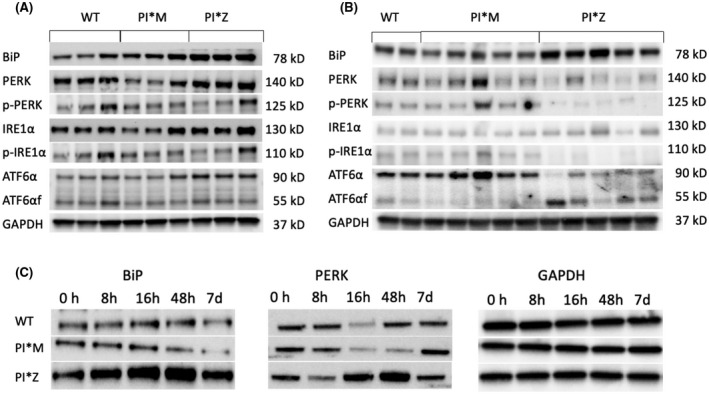
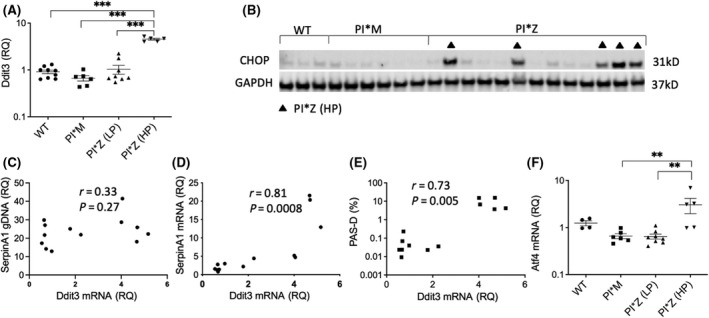
Alpha-1 antitrypsin (AAT) deficiency (AATD) is an inherited disease caused by mutations in the serpin family A member 1 (SERPINA1, also known as AAT) gene. The most common variant, PI*Z (Glu342Lys), causes accumulation of aberrantly folded AAT in the endoplasmic reticulum (ER) of hepatocytes that is associated with a toxic gain of function, hepatocellular injury, liver fibrosis, and hepatocellular carcinoma. The unfolded protein response (UPR) is a cellular response to improperly folded proteins meant to alleviate ER stress. It has been unclear whether PI*Z AAT elicits liver cell UPR, due in part to limitations of current cellular and animal models. This study investigates whether UPR is activated in a novel human PI*Z AAT cell line and a new PI*Z human AAT (hAAT) mouse model. A PI*Z AAT hepatocyte cell line (Huh7.5Z) was established using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing of the normal ATT (PI*MM) gene in the Huh7.5 cell line. Additionally, novel full-length genomic DNA PI*Z hAAT and PI*M hAAT transgenic mouse models were established. Using these new models, UPR in Huh7.5Z cells and PI*Z mice were comprehensively determined. Robust activation of UPR was observed in Huh7.5Z cells compared to Huh7.5 cells. Activated caspase cascade and apoptosis markers, increased chaperones, and autophagy markers were also detected in Z hepatocytes. Selective attenuation of UPR signaling branches was observed in PI*Z hAAT mice in which the protein kinase R-like ER kinase and inositol-requiring enzyme1α branches were suppressed while the activating transcription factor 6α branch remained active. This study provides direct evidence that PI*Z AAT triggers canonical UPR and that hepatocytes survive pro-apoptotic UPR by selective suppression of UPR branches. Our data improve understanding of underlying pathological molecular mechanisms of PI*Z AATD liver disease.
© 2022 The Authors. Hepatology Communications published by Wiley Periodicals LLC on behalf of American Association for the Study of Liver Diseases.
Conflict of interest statement
The authors declare no conflict of interest that pertain to this work.
Figures
References
-
- Huntington JA, Read RJ, Carrell RW. Structure of a serpin‐protease complex shows inhibition by deformation. Nature. 2000;407:923–6. - PubMed
-
- Brantly M, Nukiwa T, Crystal RG. Molecular basis of alpha‐1‐antitrypsin deficiency. Am J Med. 1988;84:13–31. - PubMed
-
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1‐antitrypsin accumulation in the liver. Nature. 1992;357:605–7. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
