

The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and x-ray diffraction at room temperature
- PMID: 35622909
- PMCID: PMC9140965
- DOI: 10.1126/sciadv.abo5083
The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and x-ray diffraction at room temperature
Abstract
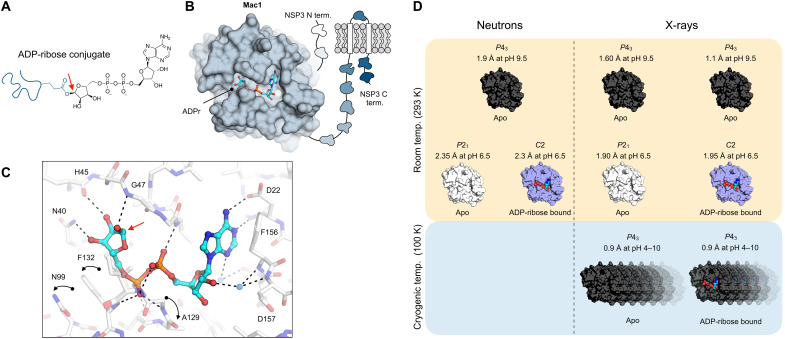
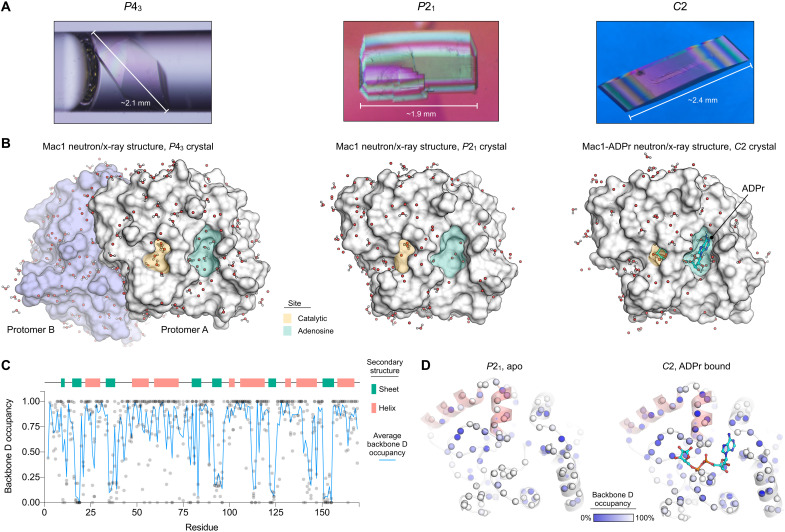
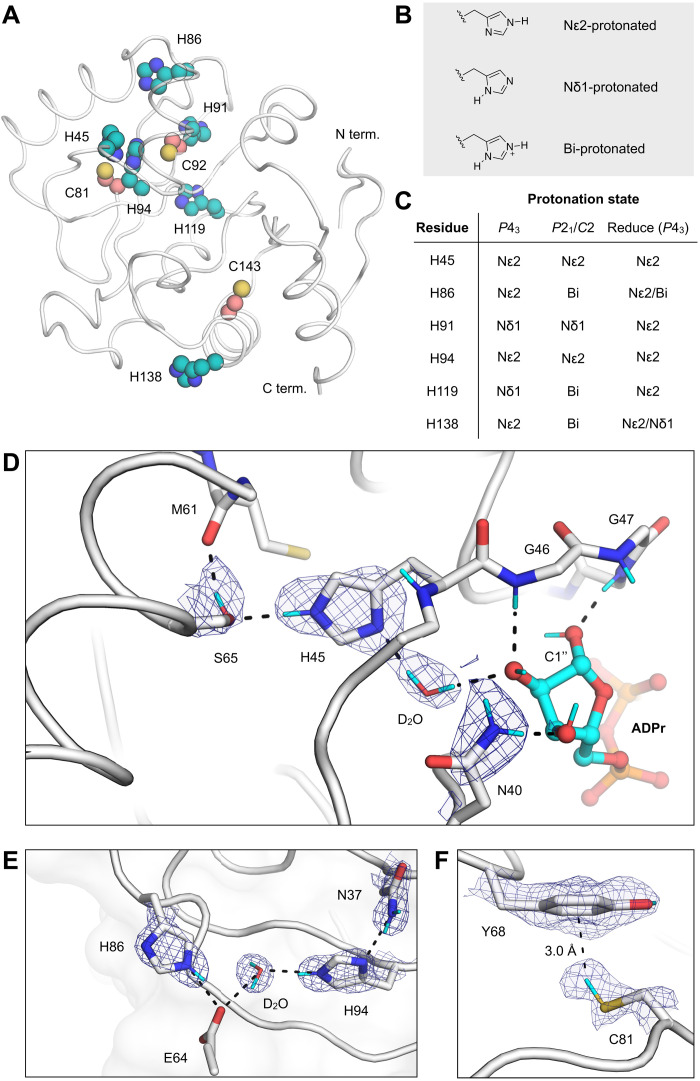
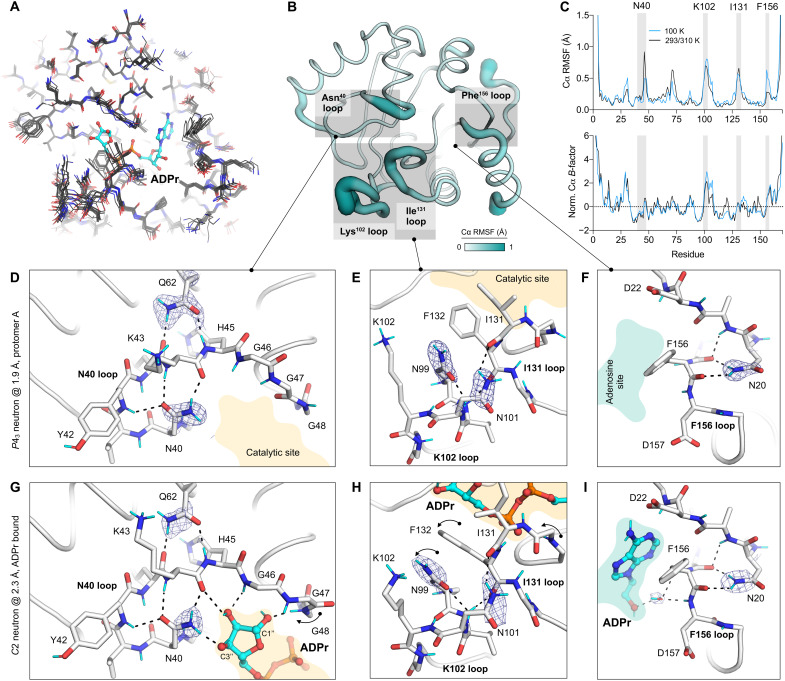
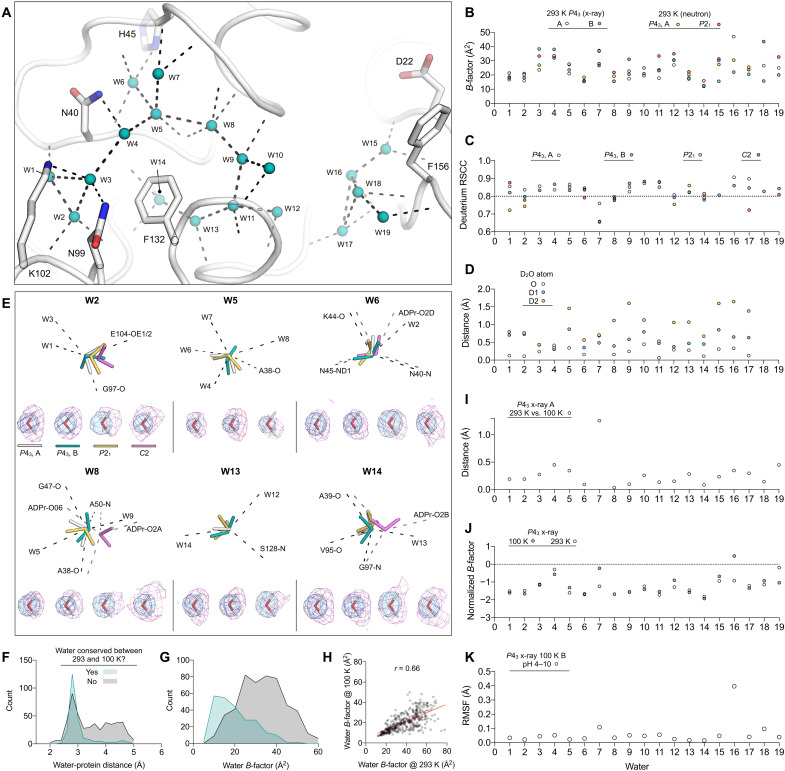
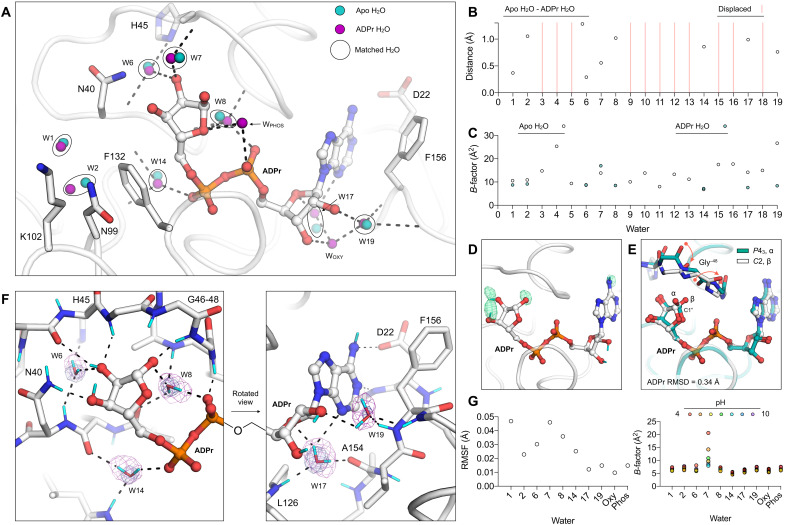
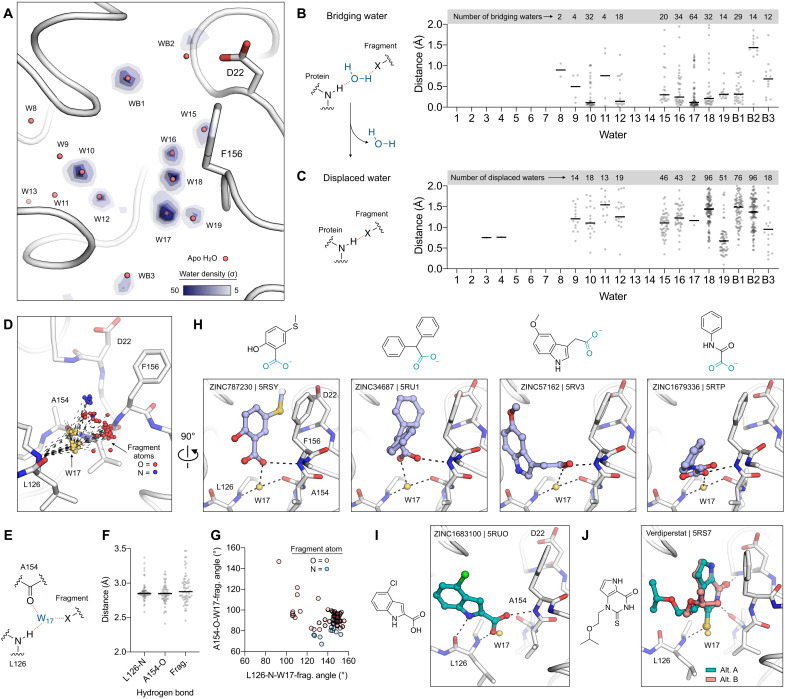
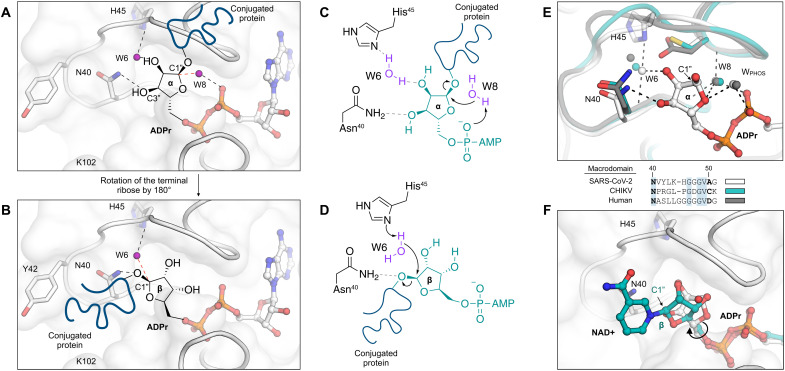
The nonstructural protein 3 (NSP3) macrodomain of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Mac1) removes adenosine diphosphate (ADP) ribosylation posttranslational modifications, playing a key role in the immune evasion capabilities of the virus responsible for the coronavirus disease 2019 pandemic. Here, we determined neutron and x-ray crystal structures of the SARS-CoV-2 NSP3 macrodomain using multiple crystal forms, temperatures, and pHs, across the apo and ADP-ribose-bound states. We characterize extensive solvation in the Mac1 active site and visualize how water networks reorganize upon binding of ADP-ribose and non-native ligands, inspiring strategies for displacing waters to increase the potency of Mac1 inhibitors. Determining the precise orientations of active site water molecules and the protonation states of key catalytic site residues by neutron crystallography suggests a catalytic mechanism for coronavirus macrodomains distinct from the substrate-assisted mechanism proposed for human MacroD2. These data provoke a reevaluation of macrodomain catalytic mechanisms and will guide the optimization of Mac1 inhibitors.
Figures
Update of
-
The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and X-ray diffraction at room temperature.bioRxiv [Preprint]. 2022 Feb 9:2022.02.07.479477. doi: 10.1101/2022.02.07.479477. bioRxiv. 2022. Update in: Sci Adv. 2022 May 27;8(21):eabo5083. doi: 10.1126/sciadv.abo5083. PMID: 35169801 Free PMC article. Updated. Preprint.
References
-
- Schoggins J. W., Interferon-stimulated genes: What do they all do? Annu Rev Virol. 6, 567–584 (2019). - PubMed
-
- Russo L. C., Tomasin R., Matos I. A., Manucci A. C., Sowa S. T., Dale K., Caldecott K. W., Lehtiö L., Schechtman D., Meotti F. C., Bruni-Cardoso A., Hoch N. C., The SARS-CoV-2 Nsp3 macrodomain reverses PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling. J. Biol. Chem. 297, 101041 (2021). - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
