

Transmission of SARS-CoV-2 from humans to animals and potential host adaptation
- PMID: 35624123
- PMCID: PMC9142586
- DOI: 10.1038/s41467-022-30698-6
Transmission of SARS-CoV-2 from humans to animals and potential host adaptation
Abstract
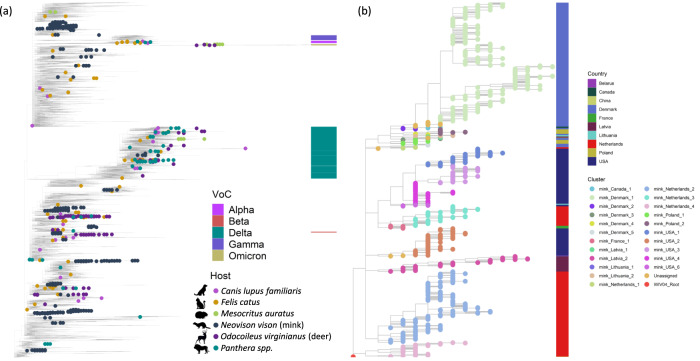
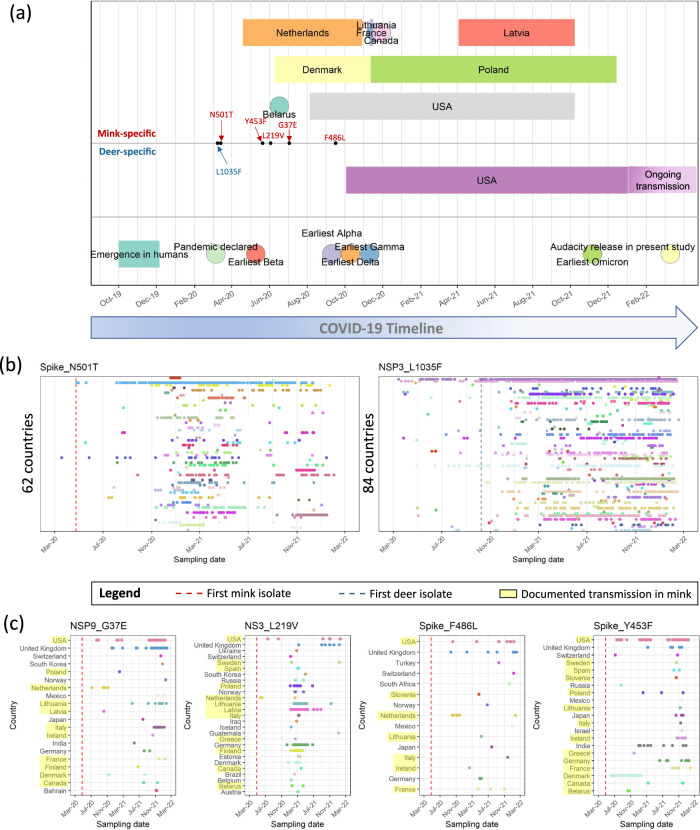
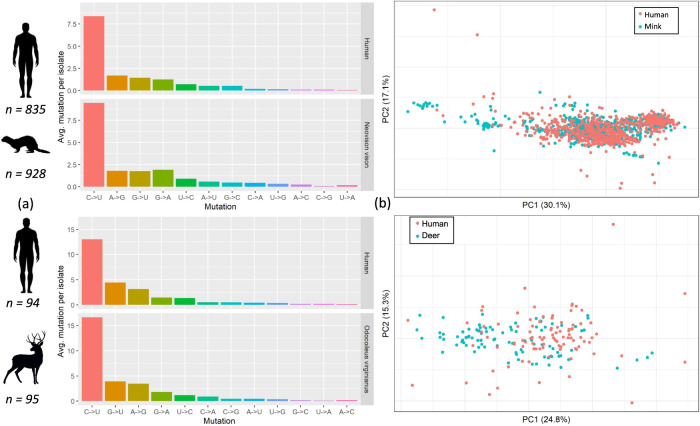
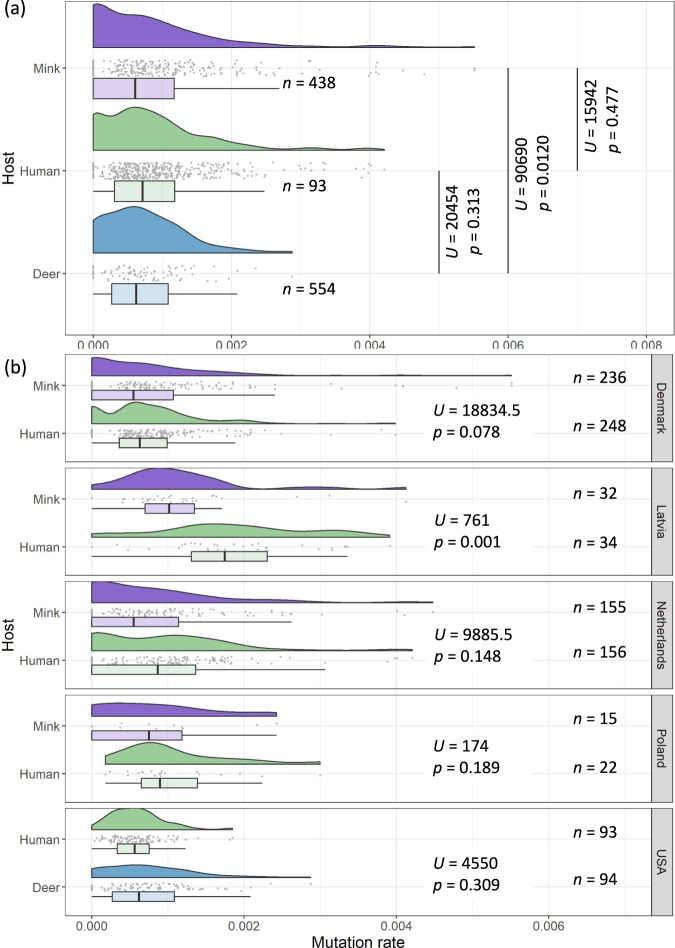
SARS-CoV-2, the causative agent of the COVID-19 pandemic, can infect a wide range of mammals. Since its spread in humans, secondary host jumps of SARS-CoV-2 from humans to multiple domestic and wild populations of mammals have been documented. Understanding the extent of adaptation to these animal hosts is critical for assessing the threat that the spillback of animal-adapted SARS-CoV-2 into humans poses. We compare the genomic landscapes of SARS-CoV-2 isolated from animal species to that in humans, profiling the mutational biases indicative of potentially different selective pressures in animals. We focus on viral genomes isolated from mink (Neovison vison) and white-tailed deer (Odocoileus virginianus) for which multiple independent outbreaks driven by onward animal-to-animal transmission have been reported. We identify five candidate mutations for animal-specific adaptation in mink (NSP9_G37E, Spike_F486L, Spike_N501T, Spike_Y453F, ORF3a_L219V), and one in deer (NSP3a_L1035F), though they appear to confer a minimal advantage for human-to-human transmission. No considerable changes to the mutation rate or evolutionary trajectory of SARS-CoV-2 has resulted from circulation in mink and deer thus far. Our findings suggest that minimal adaptation was required for onward transmission in mink and deer following human-to-animal spillover, highlighting the 'generalist' nature of SARS-CoV-2 as a mammalian pathogen.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
