

The functional evolution of termite gut microbiota
- PMID: 35624491
- PMCID: PMC9137090
- DOI: 10.1186/s40168-022-01258-3
The functional evolution of termite gut microbiota
Abstract
Background: Termites primarily feed on lignocellulose or soil in association with specific gut microbes. The functioning of the termite gut microbiota is partly understood in a handful of wood-feeding pest species but remains largely unknown in other taxa. We intend to fill this gap and provide a global understanding of the functional evolution of termite gut microbiota.
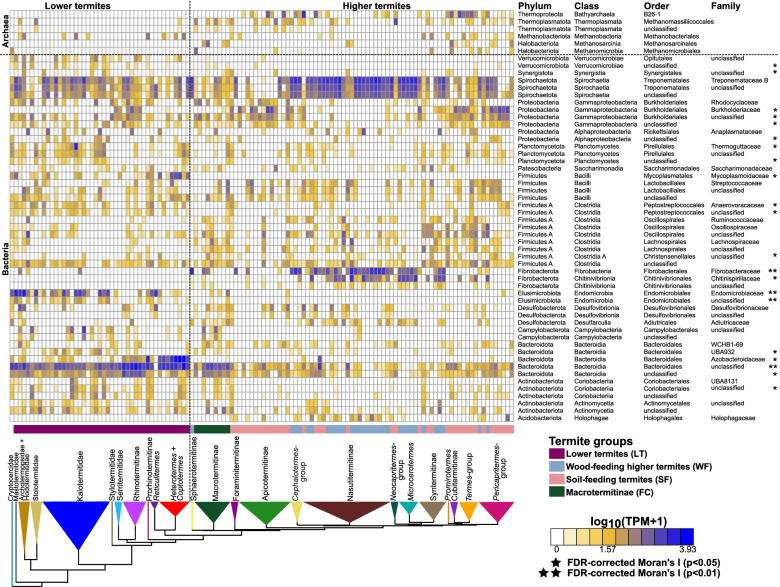
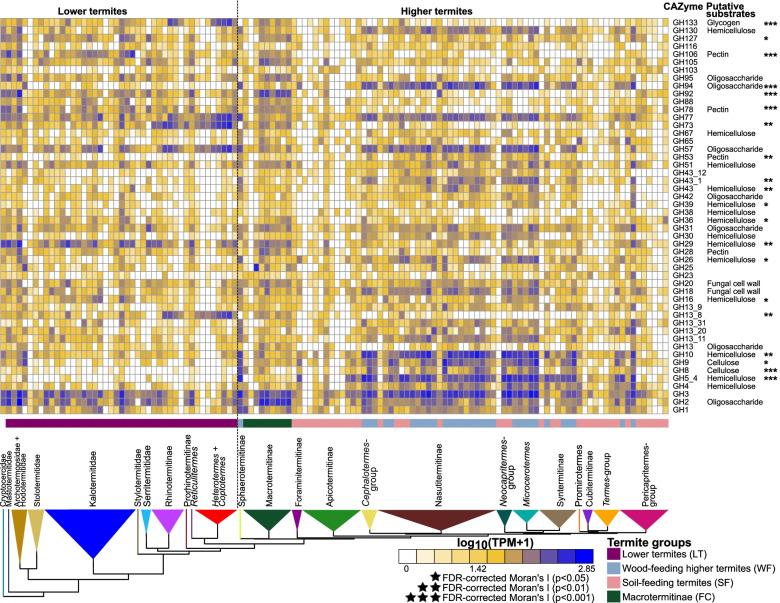
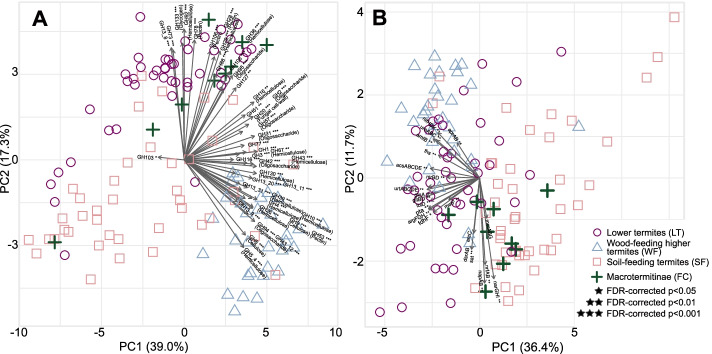
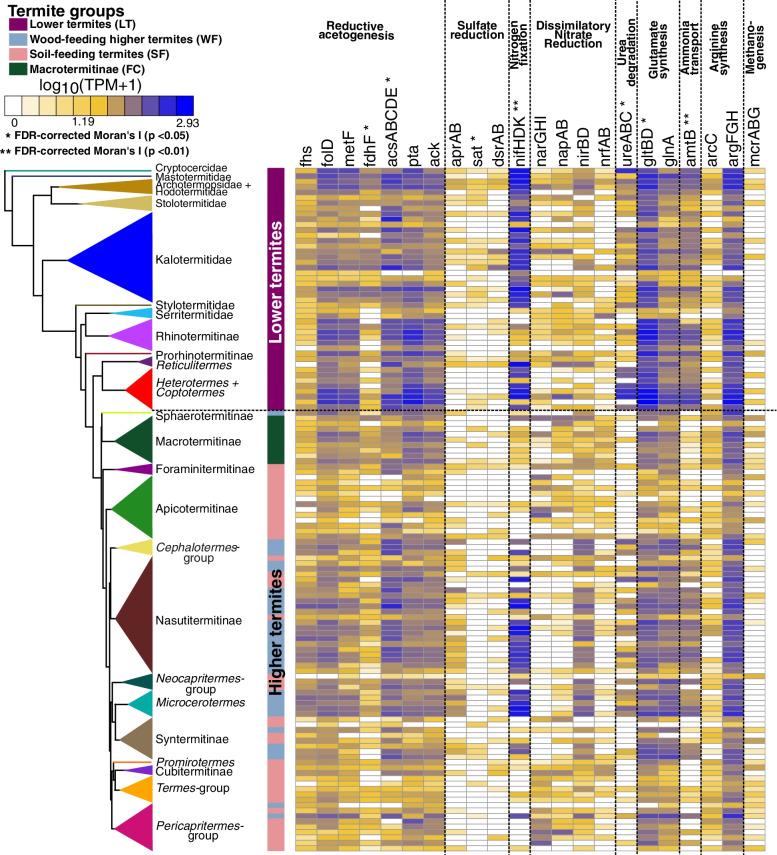
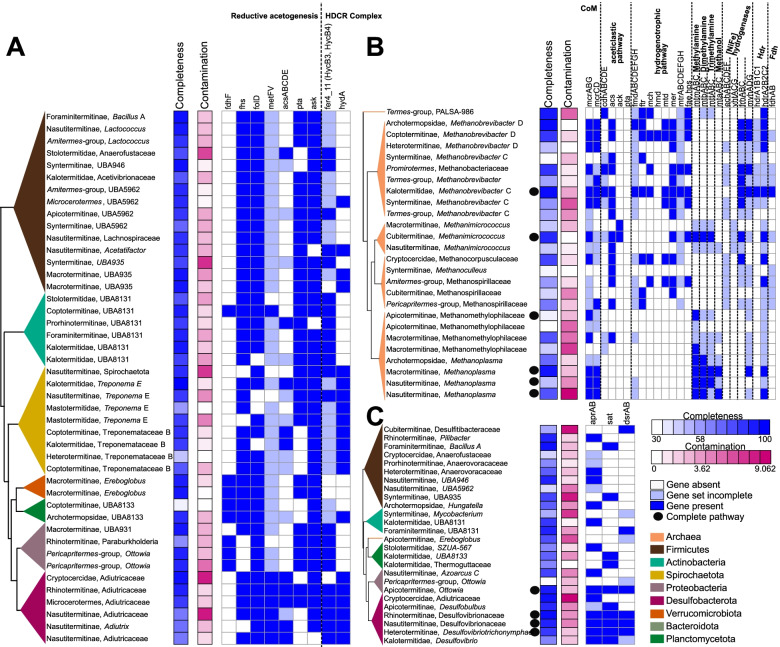
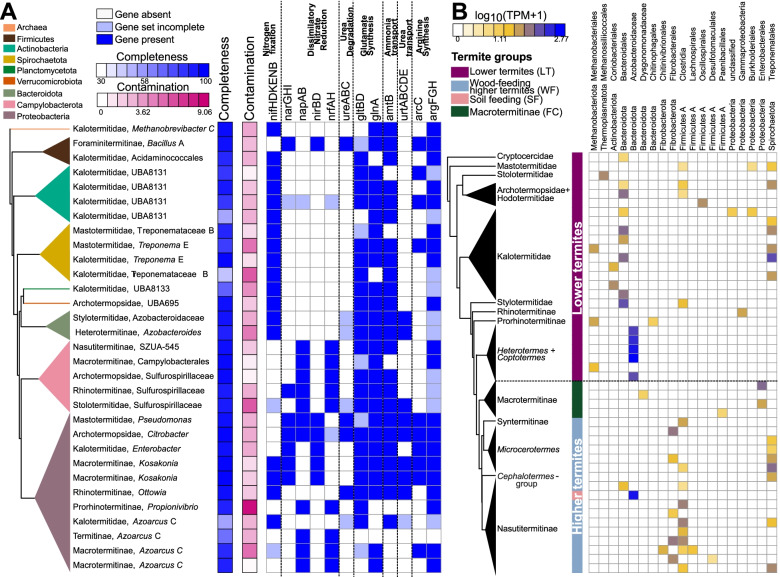
Results: We sequenced the gut metagenomes of 145 samples representative of the termite diversity. We show that the prokaryotic fraction of the gut microbiota of all termites possesses similar genes for carbohydrate and nitrogen metabolisms, in proportions varying with termite phylogenetic position and diet. The presence of a conserved set of gut prokaryotic genes implies that essential nutritional functions were present in the ancestor of modern termites. Furthermore, the abundance of these genes largely correlated with the host phylogeny. Finally, we found that the adaptation to a diet of soil by some termite lineages was accompanied by a change in the stoichiometry of genes involved in important nutritional functions rather than by the acquisition of new genes and pathways.
Conclusions: Our results reveal that the composition and function of termite gut prokaryotic communities have been remarkably conserved since termites first appeared ~ 150 million years ago. Therefore, the "world's smallest bioreactor" has been operating as a multipartite symbiosis composed of termites, archaea, bacteria, and cellulolytic flagellates since its inception. Video Abstract.
Keywords: Endosymbionts; Isoptera; Metagenomics; Vertical inheritance.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Abdul Rahman N, Parks DH, Willner DL, Engelbrektson AL, Goffredi SK, Warnecke F, Scheffrahn RH, Hugenholtz P. A molecular survey of Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome. 2015;3:5. doi: 10.1186/s40168-015-0067-8. - DOI - PMC - PubMed
-
- Adams DC, Otárola-Castillo E. Geomorph: An R package for the collection and analysis of geometric morphometric shape data. Methods Ecol Evol. 2013;4:393–399. doi: 10.1111/2041-210X.12035. - DOI
-
- Apprill A, Mcnally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol. 2015;75:129–137. doi: 10.3354/ame01753. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
