

Controlling Cancer Cell Death Types to Optimize Anti-Tumor Immunity
- PMID: 35625711
- PMCID: PMC9138898
- DOI: 10.3390/biomedicines10050974
Controlling Cancer Cell Death Types to Optimize Anti-Tumor Immunity
Abstract
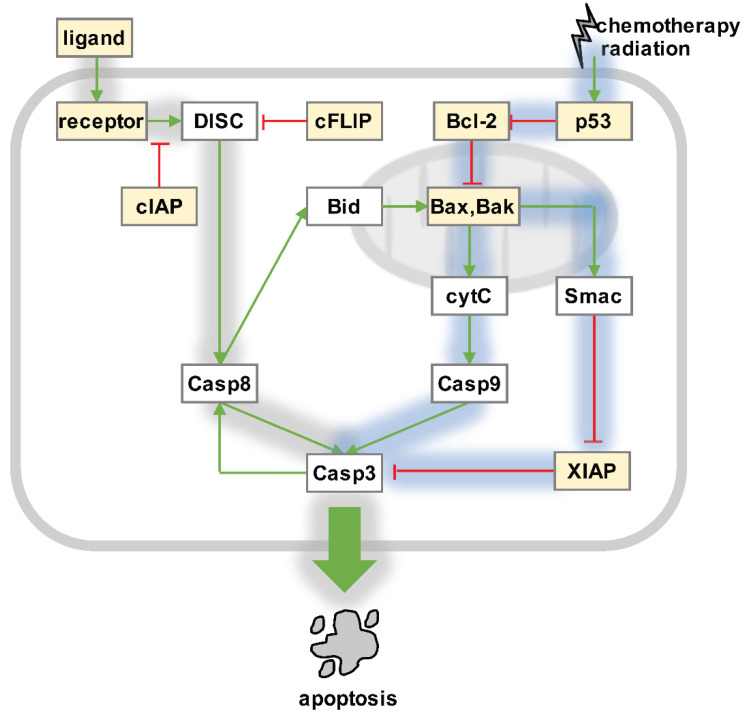
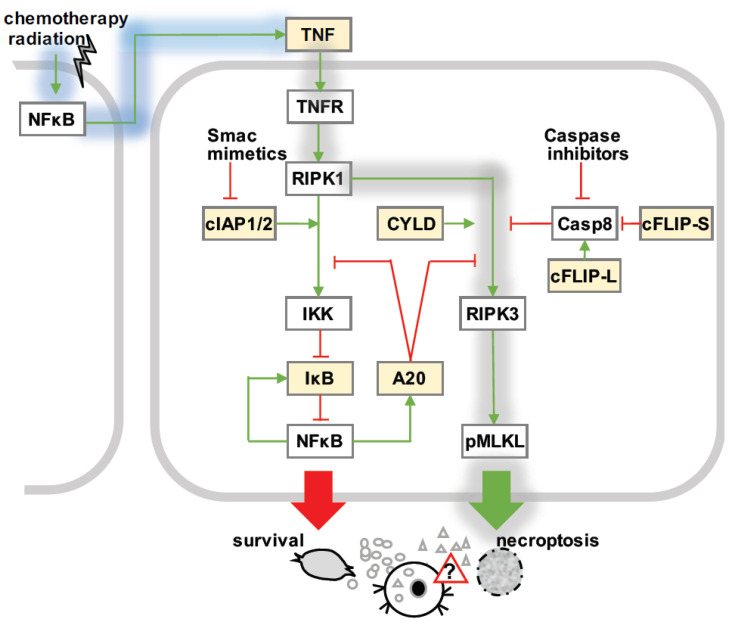
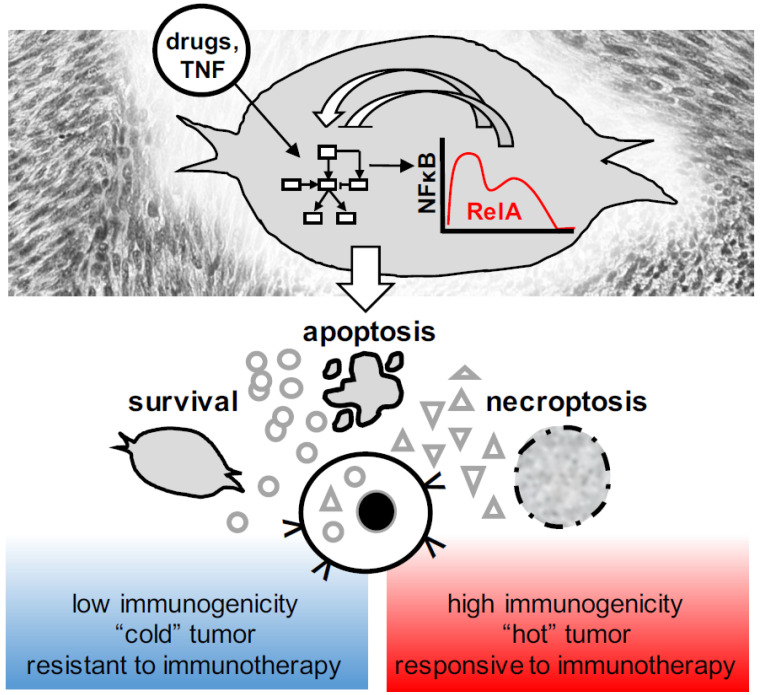
Over several decades, cell biology research has characterized distinct forms of regulated cell death, identified master regulators such as nuclear factor kappa B (NFκB), and contributed to translating these findings in order to improve anti-cancer therapies. In the era of immunotherapy, however, the field warrants a new appraisal-the targeted induction of immunogenic cell death may offer personalized strategies to optimize anti-tumor immunity. Once again, the spotlight is on NFκB, which is not only a master regulator of cancer cell death, survival, and inflammation, but also of adaptive anti-tumor immune responses that are triggered by dying tumor cells.
Keywords: NFκB dynamics; anti-tumor immunity; fate decisions; immunogenic cell death; immunotherapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation - implications for cancer therapies.Curr Med Chem. 2012;19(12):1751-64. doi: 10.2174/092986712800099811. Curr Med Chem. 2012. PMID: 22414083 Review.
-
Peripherally-driven myeloid NFkB and IFN/ISG responses predict malignancy risk, survival, and immunotherapy regime in ovarian cancer.J Immunother Cancer. 2021 Nov;9(11):e003609. doi: 10.1136/jitc-2021-003609. J Immunother Cancer. 2021. PMID: 34795003 Free PMC article.
-
Dying of Stress: Chemotherapy, Radiotherapy, and Small-Molecule Inhibitors in Immunogenic Cell Death and Immunogenic Modulation.Cells. 2022 Nov 29;11(23):3826. doi: 10.3390/cells11233826. Cells. 2022. PMID: 36497086 Free PMC article. Review.
-
Pyroptotic and Necroptotic Cell Death in the Tumor Microenvironment and Their Potential to Stimulate Anti-Tumor Immune Responses.Front Oncol. 2021 Aug 19;11:731598. doi: 10.3389/fonc.2021.731598. eCollection 2021. Front Oncol. 2021. PMID: 34490126 Free PMC article. Review.
-
Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity.J Immunother Cancer. 2020 Nov;8(2):e001369. doi: 10.1136/jitc-2020-001369. J Immunother Cancer. 2020. PMID: 33188036 Free PMC article.
References
-
- Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W., et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. - DOI - PMC - PubMed
