

doi: 10.1038/s41592-022-01488-1.
Epub 2022 May 30.
ColabFold: making protein folding accessible to all
Affiliations
- PMID: 35637307
- PMCID: PMC9184281
- DOI: 10.1038/s41592-022-01488-1
Item in Clipboard
ColabFold: making protein folding accessible to all
Nat Methods.
2022 Jun.
Abstract
ColabFold offers accelerated prediction of protein structures and complexes by combining the fast homology search of MMseqs2 with AlphaFold2 or RoseTTAFold. ColabFold's 40-60-fold faster search and optimized model utilization enables prediction of close to 1,000 structures per day on a server with one graphics processing unit. Coupled with Google Colaboratory, ColabFold becomes a free and accessible platform for protein folding. ColabFold is open-source software available at https://github.com/sokrypton/ColabFold and its novel environmental databases are available at https://colabfold.mmseqs.com .
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
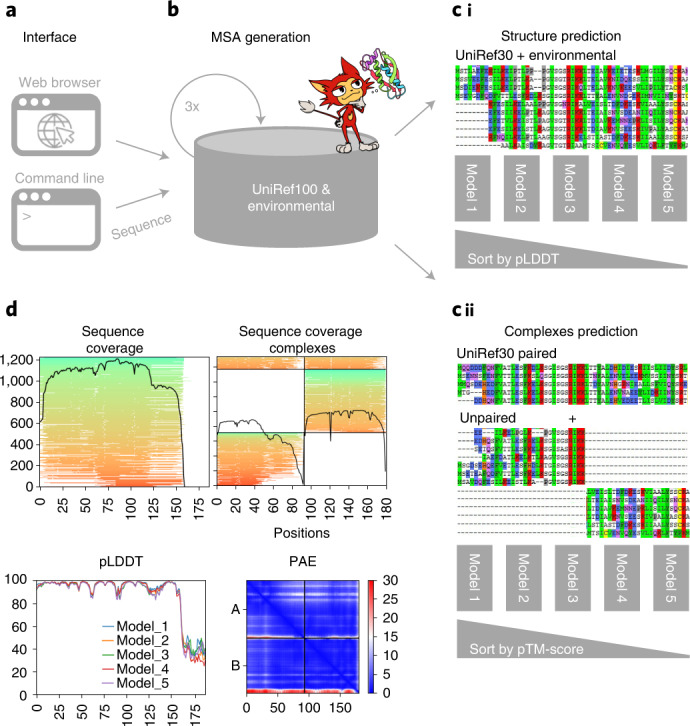
a,b, ColabFold has a web and a command line interface (a) that send FASTA input sequence(s) to an MMseqs2 server (b) searching two databases, UniRef100 and a database of environmental sequences, with three profile-search iterations each. The second database is searched using a sequence profile generated from the UniRef100 search as input. The server generates two MSAs in A3M format containing all detected sequences. c, For predictions of single structures (i) we filter both A3Ms using a diversity-aware filter and return this to be provided as the MSA input feature to the AlphaFold2 models. For predictions of complexes (ii) we pair the top hits within the same species to resolve the inter-chain contacts and additionally add two unpaired MSAs (same as i) to guide the structure prediction. Single chain predictions are ranked by pLDDT and complexes by predicted TM-score. d, To help researchers judge the prediction quality we visualize MSA depth and diversity and show the AlphaFold2 confidence measures (pLDDT and PAE).
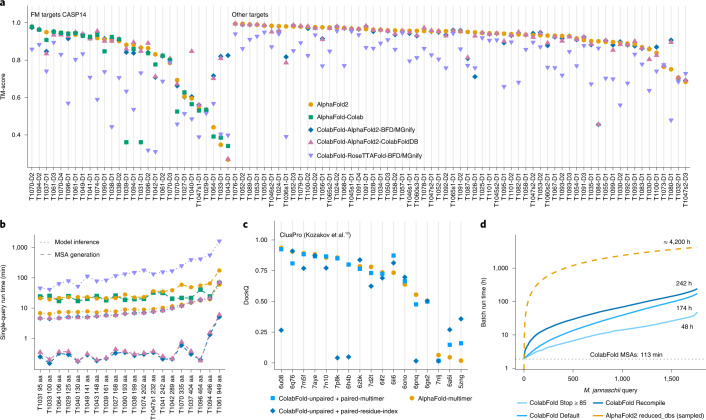
a, Structure prediction comparison of AlphaFold2, AlphaFold-Colab and ColabFold-AlphaFold2 with BFD/MGnify and with the ColabFoldDB, and ColabFold-RoseTTAFold with BFD/MGnify using predictions of 91 domains of 65 CASP14 targets. The 28 domains from the 20 free-modeling (FM) targets are shown first. FM targets were used to optimize MMseqs2 search parameters. Each target was evaluated for each individual domain (in total 91 domains). b, MSA generation and model inference times for each CASP14 FM target sorted by protein length (same colors as before). Blue shows MSA run times for ColabFold-AlphaFold2-BFD/MGnify and ColabFold-RoseTTAFold-BFD/MGnify. c, Comparison of multimeric prediction modes in ColabFold and AlphaFold-multimer. The ColabFold modes include residue-index modification with models originally trained for single-chain predictions and those for multimeric prediction from AlphaFold-multimer, using DockQ (a quality measure for protein–protein docking models). d, Run time of colabfold_batch proteome prediction at three optimization levels: always recompile, default, and stop model/recycle evaluation after first prediction with a pLDDT of ≥85. The yellow dashed line represents an extrapolation on the basis of the 50 AlphaFold2 predictions. Source data
References
-
- Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv10.1101/2021.10.04.463034 (2021).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
