

Polycomb repressive complex 2 shields naïve human pluripotent cells from trophectoderm differentiation
- PMID: 35637409
- PMCID: PMC9203276
- DOI: 10.1038/s41556-022-00916-w
Polycomb repressive complex 2 shields naïve human pluripotent cells from trophectoderm differentiation
Abstract
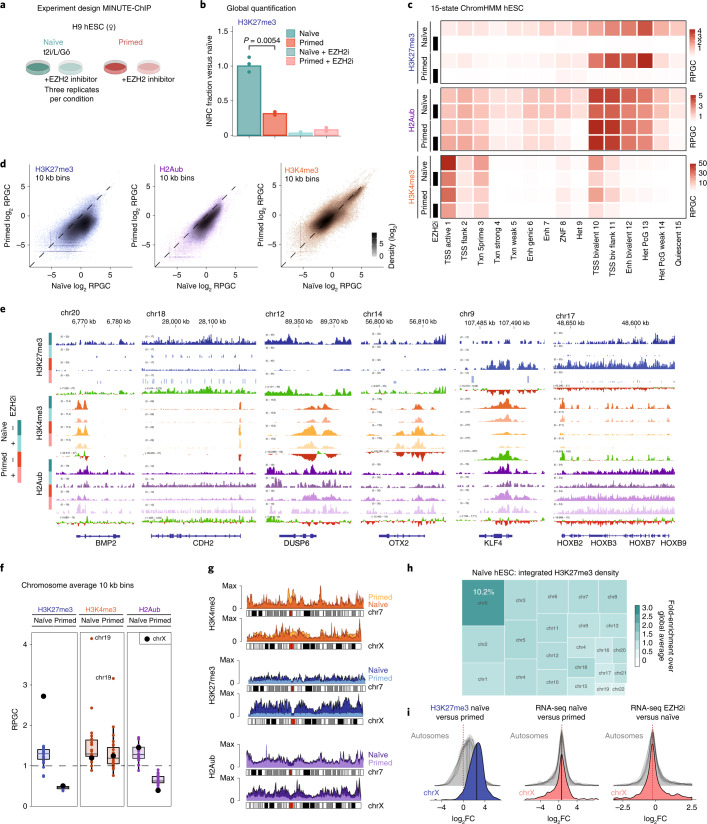
The first lineage choice in human embryo development separates trophectoderm from the inner cell mass. Naïve human embryonic stem cells are derived from the inner cell mass and offer possibilities to explore how lineage integrity is maintained. Here, we discover that polycomb repressive complex 2 (PRC2) maintains naïve pluripotency and restricts differentiation to trophectoderm and mesoderm lineages. Through quantitative epigenome profiling, we found that a broad gain of histone H3 lysine 27 trimethylation (H3K27me3) is a distinct feature of naïve pluripotency. We define shared and naïve-specific bivalent promoters featuring PRC2-mediated H3K27me3 concomitant with H3K4me3. Naïve bivalency maintains key trophectoderm and mesoderm transcription factors in a transcriptionally poised state. Inhibition of PRC2 forces naïve human embryonic stem cells into an 'activated' state, characterized by co-expression of pluripotency and lineage-specific transcription factors, followed by differentiation into either trophectoderm or mesoderm lineages. In summary, PRC2-mediated repression provides a highly adaptive mechanism to restrict lineage potential during early human development.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures














Comment in
-
PRC2 shields the potency of human stem cells.Nat Cell Biol. 2022 Jun;24(6):806-808. doi: 10.1038/s41556-022-00937-5. Nat Cell Biol. 2022. PMID: 35697784 No abstract available.
References
-
- Thomson, J. A. et al. Embryonic stem cell lines derived from human blastocysts. Science282, 1145–1147 (1998). - PubMed
-
- Xu, R.-H. et al. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol.20, 1261–1264 (2002). - PubMed
-
- Marchand, M. et al. Transcriptomic signature of trophoblast differentiation in a human embryonic stem cell model. Biol. Reprod.84, 1258–1271 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
