

APOLD1 loss causes endothelial dysfunction involving cell junctions, cytoskeletal architecture, and Weibel-Palade bodies, while disrupting hemostasis
- PMID: 35638551
- PMCID: PMC9973481
- DOI: 10.3324/haematol.2022.280816
APOLD1 loss causes endothelial dysfunction involving cell junctions, cytoskeletal architecture, and Weibel-Palade bodies, while disrupting hemostasis
Abstract
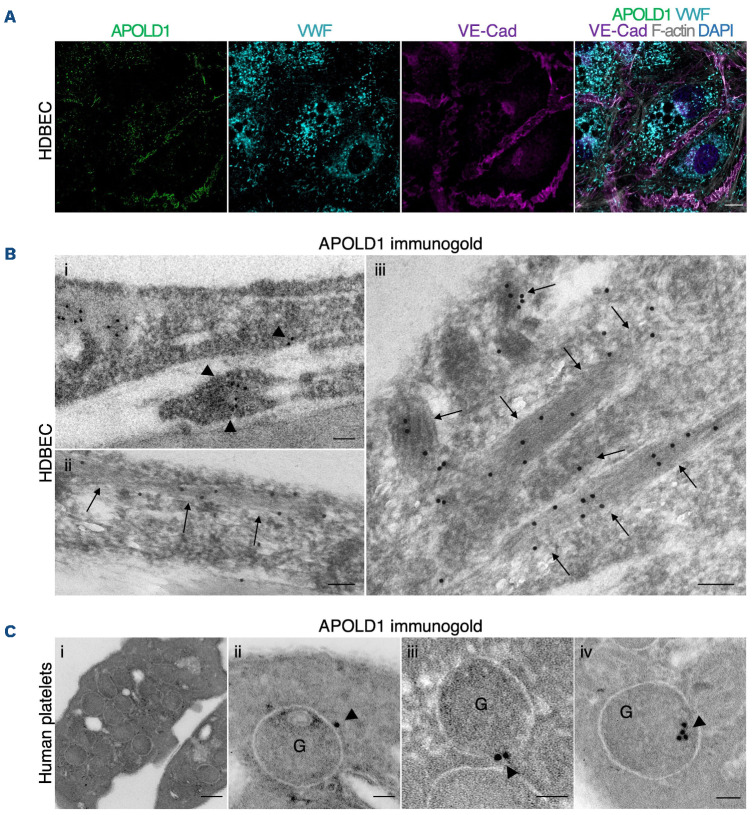
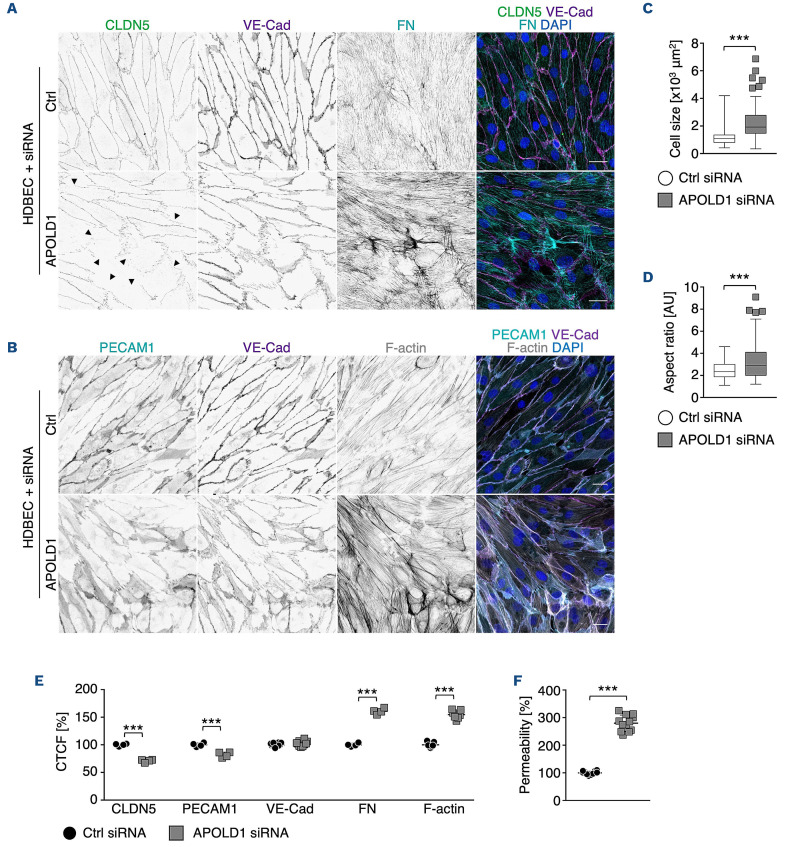
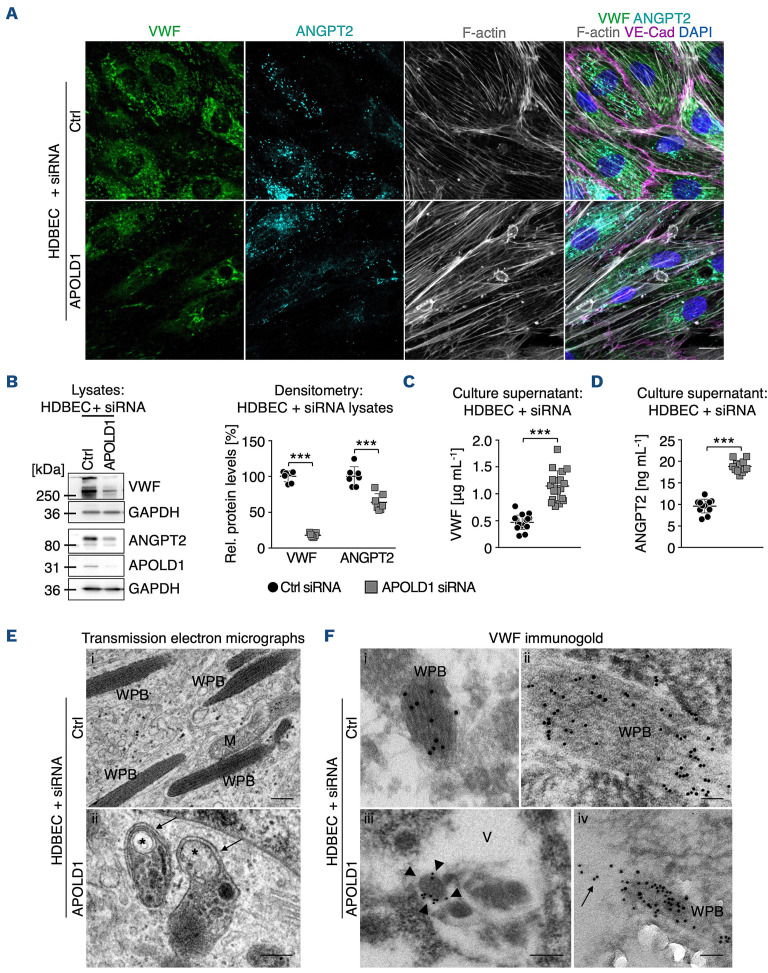
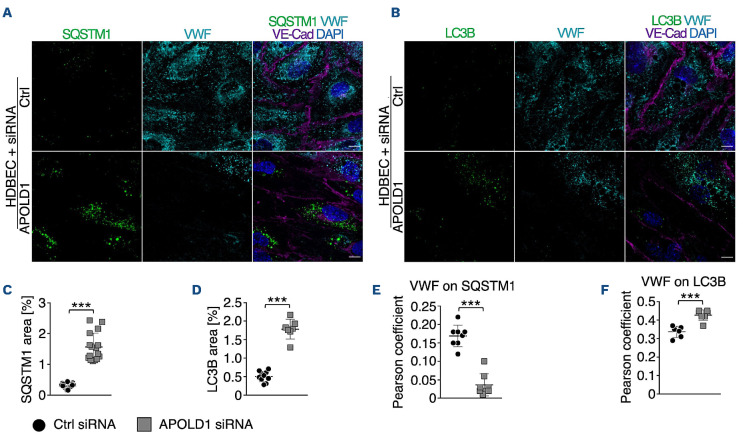
Vascular homeostasis is impaired in various diseases thereby contributing to the progression of their underlying pathologies. The endothelial immediate early gene Apolipoprotein L domain-containing 1 (APOLD1) helps to regulate endothelial function. However, its precise role in endothelial cell biology remains unclear. We have localized APOLD1 to endothelial cell contacts and to Weibel-Palade bodies (WPB) where it associates with von Willebrand factor (VWF) tubules. Silencing of APOLD1 in primary human endothelial cells disrupted the cell junction-cytoskeletal interface, thereby altering endothelial permeability accompanied by spontaneous release of WPB contents. This resulted in an increased presence of WPB cargoes, notably VWF and angiopoietin-2 in the extracellular medium. Autophagy flux, previously recognized as an essential mechanism for the regulated release of WPB, was impaired in the absence of APOLD1. In addition, we report APOLD1 as a candidate gene for a novel inherited bleeding disorder across three generations of a large family in which an atypical bleeding diathesis was associated with episodic impaired microcirculation. A dominant heterozygous nonsense APOLD1:p.R49* variant segregated to affected family members. Compromised vascular integrity resulting from an excess of plasma angiopoietin-2, and locally impaired availability of VWF may explain the unusual clinical profile of APOLD1:p.R49* patients. In summary, our findings identify APOLD1 as an important regulator of vascular homeostasis and raise the need to consider testing of endothelial cell function in patients with inherited bleeding disorders without apparent platelet or coagulation defects.
Figures


Comment in
-
Loss of APOLD1: a new vascular bleeding disorder?Haematologica. 2023 Mar 1;108(3):665-667. doi: 10.3324/haematol.2022.281354. Haematologica. 2023. PMID: 35638552 Free PMC article. No abstract available.
References
-
- Bogatcheva NV, Garcia JGN, Verin AD. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry (Mosc). 2002;67(1):75-84. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
