

Heterozygous PRKN mutations are common but do not increase the risk of Parkinson's disease
- PMID: 35640906
- PMCID: PMC9423714
- DOI: 10.1093/brain/awab456
Heterozygous PRKN mutations are common but do not increase the risk of Parkinson's disease
Abstract
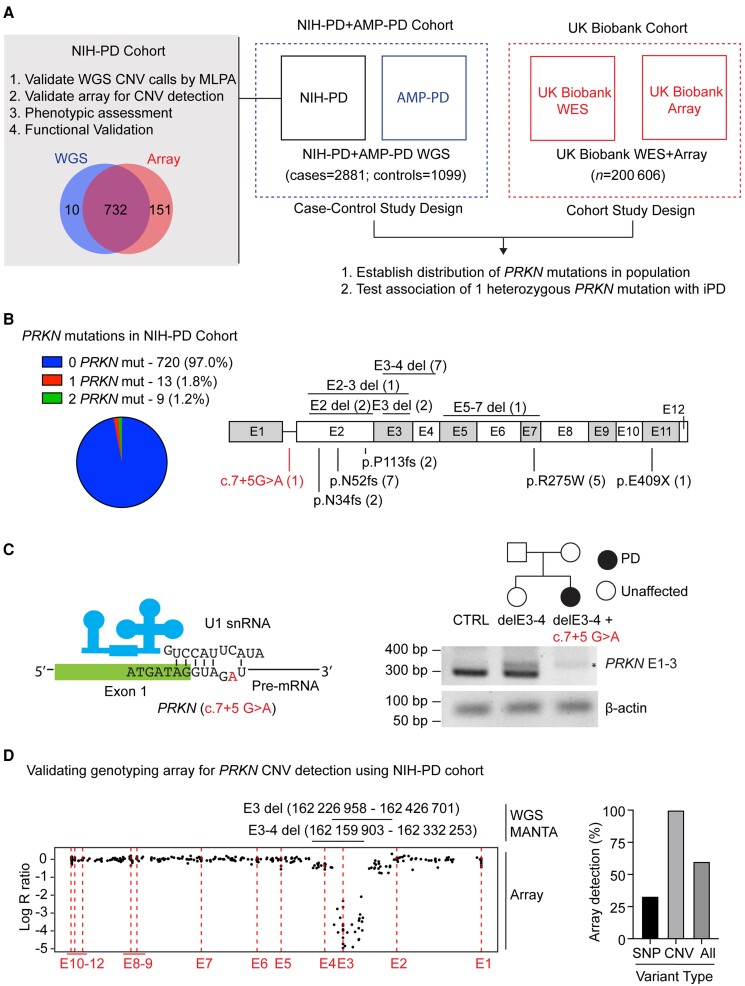
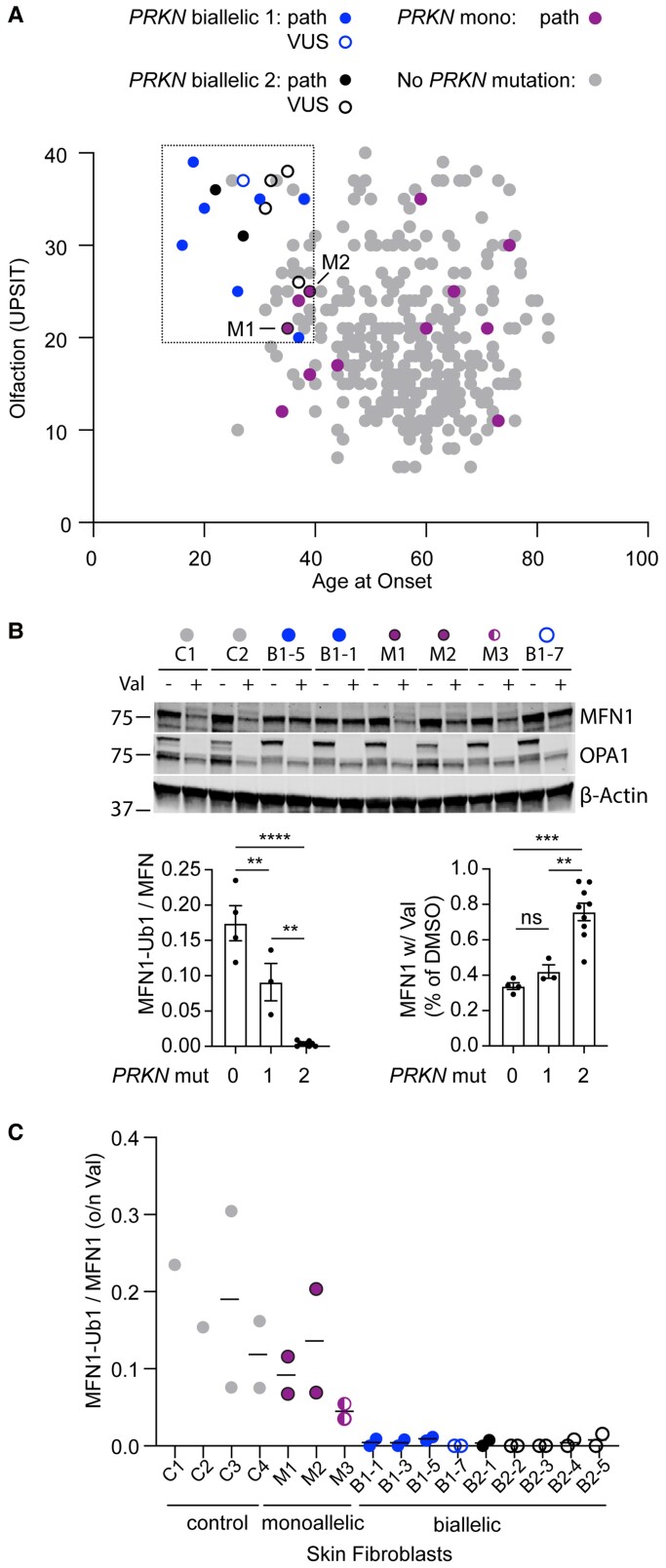
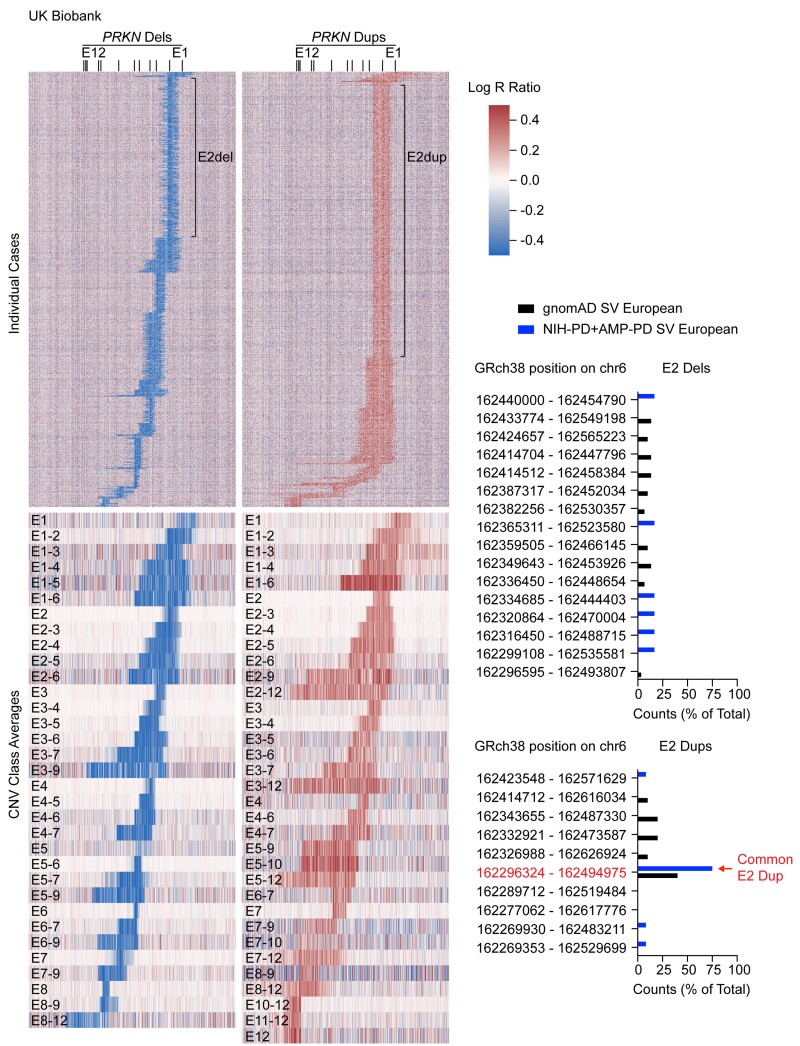
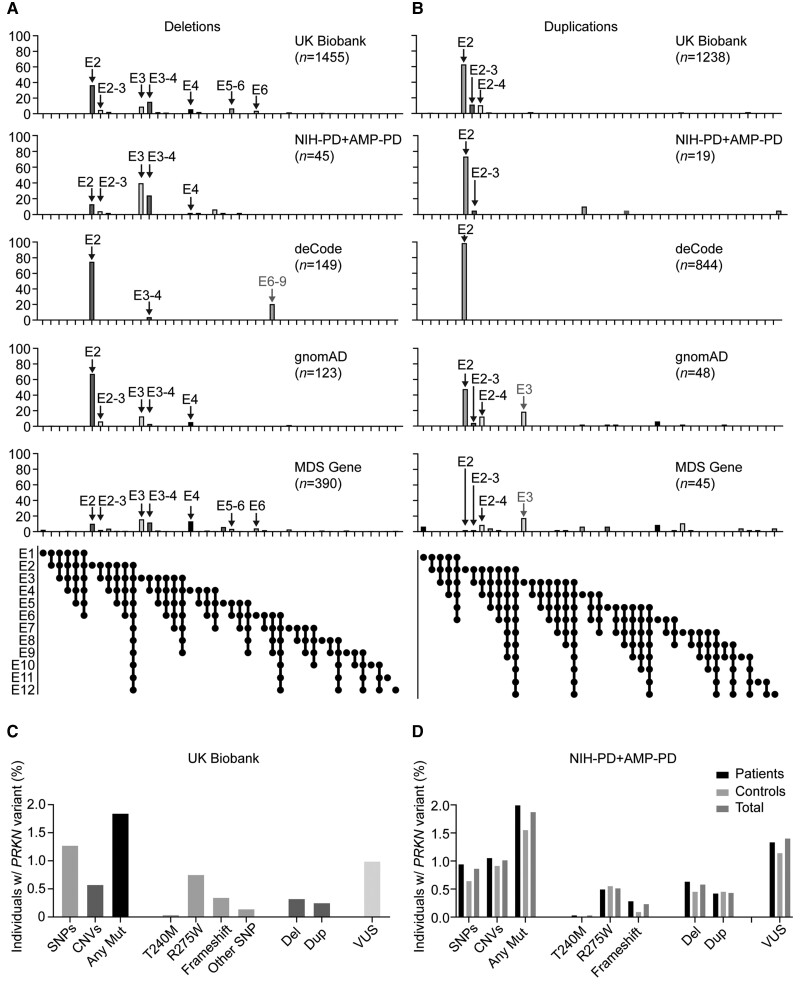
PRKN mutations are the most common recessive cause of Parkinson's disease and are a promising target for gene and cell replacement therapies. Identification of biallelic PRKN patients at the population scale, however, remains a challenge, as roughly half are copy number variants and many single nucleotide polymorphisms are of unclear significance. Additionally, the true prevalence and disease risk associated with heterozygous PRKN mutations is unclear, as a comprehensive assessment of PRKN mutations has not been performed at a population scale. To address these challenges, we evaluated PRKN mutations in two cohorts with near complete genotyping of both single nucleotide polymorphisms and copy number variants: the NIH-PD + AMP-PD cohort, the largest Parkinson's disease case-control cohort with whole genome sequencing data from 4094 participants, and the UK Biobank, the largest cohort study with whole exome sequencing and genotyping array data from 200 606 participants. Using the NIH-PD participants, who were genotyped using whole genome sequencing, genotyping array, and multi-plex ligation-dependent probe amplification, we validated genotyping array for the detection of copy number variants. Additionally, in the NIH-PD cohort, functional assays of patient fibroblasts resolved variants of unclear significance in biallelic carriers and suggested that cryptic loss of function variants in monoallelic carriers are not a substantial confounder for association studies. In the UK Biobank, we identified 2692 PRKN copy number variants from genotyping array data from nearly half a million participants (the largest collection to date). Deletions or duplications involving exon 2 accounted for roughly half of all copy number variants and the vast majority (88%) involved exons 2, 3, or 4. In the UK Biobank, we found a pathogenic PRKN mutation in 1.8% of participants and two mutations in ∼1/7800 participants. Those with one PRKN pathogenic variant were as likely as non-carriers to have Parkinson's disease [odds ratio = 0.91 (0.58-1.38), P-value 0.76] or a parent with Parkinson's disease [odds ratio = 1.12 (0.94-1.31), P-value = 0.19]. Similarly, those in the NIH-PD + AMP + PD cohort with one PRKN pathogenic variant were as likely as non-carriers to have Parkinson's disease [odds ratio = 1.29 (0.74-2.38), P-value = 0.43]. Together our results demonstrate that heterozygous pathogenic PRKN mutations are common in the population but do not increase the risk of Parkinson's disease.
Keywords: PARK2; early onset Parkinson’s disease; mitophagy; parkin; young onset Parkinson’s disease.
Published by Oxford University Press on behalf of the Guarantors of Brain 2022. This work is written by (a) US Government employee(s) and is in the public domain in the US.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
